
簡介
 侏儒
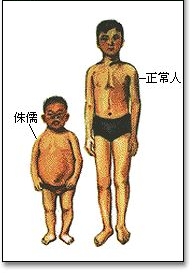
侏儒 侏儒症是由一種基因疾病引起的,會導致短小的身材和骨骼不成比例的生長。根據美國小人公司這個有5,000名成員的組織(該組織對身高4英尺10英寸以下的人開放),每年有400名嚴重的侏儒症兒童由正常身高的父母生下。儘管任何一對夫妻都有可能生下一個侏儒的孩子,但侏儒夫妻有80%的機會會有和他們一樣症狀的後代。伴隨的侏儒的症狀對不同的,但很多人另外還患有心臟和呼吸系統疾病,這會顯著地縮短他們的壽命。還有的人有異常或太小的內臟器官,這會使長期的生存變得困難。
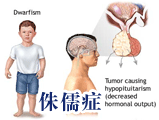
垂體一旦有病引起的侏儒症,稱為垂體侏儒。垂體侏儒的病因有兩種,一種是原發性,病因不明,部分屬遺傳性疾病。一種是繼發性,即由於垂體周圍組織有各種病變,包括腫瘤,如顱咽管瘤、垂體黃色瘤等;感染如腦炎 、 腦膜炎 、 結核病 ;血管病變及外傷。
原發性垂體侏儒多見於男孩,初生時正常,1-2歲左右發育也正常,一般3-4歲開始發現生長發育落後,隨著年齡的增長,孩子越大越顯出智力的落後。如果是繼發性的,發病年齡可在任何時候。如繼發於垂體腫瘤,症狀發生於腫瘤初起之時,並可伴有其他腫瘤的表現。垂體侏儒患兒從外觀上看,比其實際年齡要小,但其四肢、軀幹、頭面部的比例都很勻稱。只是個兒矮,整個兒成比例的縮小,智力發育可不受影響,看起來像小大人。這種孩子出牙也晚,多數有性腺發育不全,第二性徵發育不全或缺乏,往往在青春發育期後仍保持兒童面容,嗓音不變粗,仍保持音調較高的童音。 真正的垂體侏儒比較少見。因此,不要把個子矮的孩子都認為是這種病。
 侏儒症
侏儒症個子矮稱為侏儒狀態,原因很多。許多兒童期的慢 性病 都可以是它的病因,如先天性心臟病 、心肌病、慢性腎炎 、重症佝僂病、慢性 營養不良 、慢性寄生蟲病、慢性肝病(慢性 肝炎 、肝硬變)和結核病。如果有以上慢性疾病應著重治療它。病因一旦去除,就會自然長高。此外,還有一種稱為青春發育延遲症(體質性生長延遲症)是最常見的一類。不少家長為自己的孩子找 醫生 看病,大部分屬於這一類。通俗地說就是晚發育,即比正常兒童落後2-4年,年齡已到青春發育期,但仍持續矮小狀態,牙也出得晚,性發育也較遲,智力發育正常。由於內分泌無異常,早晚會發育的,只是比正常晚些,一旦達到發育期則會很快長起來,最後終歸能達到正常高度。因此,不要把這類孩子誤認為是垂體侏儒。 垂體侏儒的治療取決於病因。如果是繼發性的,應找到原發病進行治療。如果是腫瘤,需要根據情況進行手術治療。如果是炎症,應進行消炎治療,使用合適的抗生素。真正的原發性垂體侏儒,需用激素治療。目前已用生物工程生產出人工製造的生長激素,垂體侏儒患兒服用生長激素後會有立竿見影的顯著效果,但目前價格昂貴,病兒必須有一定經濟基礎否則負擔不起。
基因
 袖珍夫婦婚禮
袖珍夫婦婚禮巴西和美國科學家經過多年的研究,發現了導致侏儒症的基因。這一重大發現將有可能為醫治 增高帶來希望。
巴西東北部塞爾希培州的伊塔巴亞尼那市的居民中很大一部分患有侏儒症,這一奇怪現象引起巴西和美國科學家的注意。美國著名內分泌學家麥可·萊溫尼在巴西聖保羅大學和塞爾希培聯邦大學研究人員的幫助下,幾年前開始對這一現象進行研究,並通過DNA分析,終於分離出導致侏儒症的基因,並發現了基因的缺陷。
萊溫尼表示,這一發現有助於在產前診斷患有侏儒症的胎兒,並找到合適的基因治療方法使之痊癒。但萊溫尼同時承認,在目前的技術水平下將正常基因植入人體細胞非常困難。目前,科學家們正在研究一種能促使侏儒症患者增加生長激素分泌的口服藥,這種方法更為安全可靠。
分類
根據病因可將本病分為兩類。
 最著名的侏儒
最著名的侏儒 一、原發性多數患者原因不明,也無家族史,僅小部分有家族性發病史,為常染色體隱性遺傳。
二、繼發性較為少見,任何病變損傷垂體前葉或下丘腦時可引起生長發育停滯,常見者有腫瘤(如顱咽管瘤、視交叉或下丘腦的膠質瘤、垂體黃色瘤等),感染(如腦炎,結核、血吸蟲病、弓漿蟲病等),外傷,血管壞死及X線損傷等。
臨床表現
 侏儒症
侏儒症為內分泌性侏儒。由於先天的(原發的)或後天的(繼發的)原因,垂體前葉功能減退,生長激素分泌不足,阻礙身體生長與發育。垂體性侏儒的主要臨床表現有:1)生長遲緩,無青春發動期,成年後有“老小孩”早衰形貌;2)身體矮小,但軀幹、四肢和頭部比例對稱33)骨骺發育遲緩,骨齡低於其實際年齡;4)性器官發育不良,第二性徵不發育;5)蝶鞍可以正常(原發性垂體侏儒)或增大(由腫瘤引起的繼發性垂體侏儒);6)實驗室檢查除生長激素分泌不足外,促性腺激素常缺乏,其他幾種垂體前葉促激素的分泌量可正常或偏低。
童年各種慢性感染、全身性疾病、嚴重營養不良等,可引起中樞神經系統功能紊亂,導致全身營養代謝紊亂性侏儒症。此類侏儒症雖各有其病因和臨床表現,但都與垂體分泌功能減退有關,所表現的侏儒症狀也相似。
2、克汀病
甲狀腺激素與骨的成熟有密切關係。兒童時期甲狀腺能低下時,軟骨的骨化與牙齒的生長受阻,各長骨骨化中心出現的時間顯著推遲,影響骨路的發育和成熟,從而導致身材矮小。本症的臨床特點為:1)臉部外型發育受阻,嬰兒面容,舌大常伸口外;2)股骨、脛骨等主要長骨發育障礙,因而身體比例呈下半身短,上半身長;3)性發育較遲,或不受影響;4)智慧型低下。
克汀病又稱呆小病,其甲狀腺功能減退系發生於胎兒或嬰兒期。若甲狀腺功能減退發生時間較後,則稱作粘液性水腫。
3、軟骨發育障礙
本病由於軟骨生長能力很弱,骨路縱長發育遲緩,形成嚴重矮小畸形,有家族遺傳性,不屬內分泌性。臨床表現特點有:1)軀幹骨長度往往正常,而四肢明顯短小,下半身顯著比上半身短;2)脊柱前凸,腹部前挺,臀部後蹶,手指粗短;3)智力發育正常;4)性器官正常,有生殖能力。
4、卵巢發育不全症
 侏儒症
侏儒症本病又稱Turner綜合徵,是一種女性先天性遺傳缺陷症。患者卵巢發育不全,伴有侏儒症。其特點是:1)越接近青春期生長發育越遲緩;2)生殖器官發育不全,原發性閉經;3)身體矮胖,有盾甲狀胸;4)患者尿中促性腺激素顯著增多,這與垂體性侏儒症恰好相反。
此外,還有原基性侏儒,性早熟性矮小,均屬體質性的,而非病理性的,與遺傳有關。
診斷要點
1、出生時身長體重往往正常,1~3歲後和同齡兒童差別愈見顯著,成年時身高常在130cm以下,但身體比例、智力比例、智 侏儒症
侏儒症2、青春期第二性徵缺如,無腋、陰毛、男性無睪丸、陰莖和前列腺不發育或發育較差,女性無月經,乳房、子宮、卵巢和外陰均不發育。
3、X線攝片可見骨齡幼稚,骨骼久不融合。
4、空腹血漿生長抑制水平低於正常,且對胰島素或精氨酸的刺激反應不敏感。大部分患者可伴促性腺激素及性激素水平低下。
輔助檢查項
 侏儒症
侏儒症2、夜間12h生長激素刺激試驗示脈衝減少。
3、生長激素釋放激素試驗鑑別垂體和下丘腦病變。
4、生長介質測定常低於正常。
5、血T3、T4、TSH測定多數正常或低值。
6、骨齡測定常呈延遲。
7、女性作染色體檢查排除Turner綜合症。
8、顱腦占位性病變可作頭顱X線攝片和CT顯象。
治療方法
 侏儒症
侏儒症(1)生長激素:過去套用從人垂體中提取的生長激素,但後來發現它能引起一種病,所以現已中止使用。現在在臨床上套用的是重組人生長激素,效果良好,治療期間身高增長速度可由治療前每年不足3厘米增加到平均 10厘米以上。如治療年齡早,長期套用,可望使身高接近正常人水平。劑量為每周0.5-0.7國際單位/公斤體重,分6-7天於晚8時或睡前注射。有條件者應長期治療,直至生長基本停止。此藥價格較貴,目前尚無國產,推廣使用受一定限制。
(2)苯丙酸諾龍:這是一種人工合成的激素類藥物,具有促進蛋白質合成的作用,而雄激素作用較弱,用藥後食慾會增加,身高可以增長。一般在10-14歲開始套用,治療後半年至1年內往往效果較顯著,一般身高可增加5-10厘米。治療2-3年後,生長逐漸減慢,骨骼定形後,身高不再增加,最終身高仍明顯低於正常,效果不甚理想。在沒有生長激素的情況下可以使用。劑量為每月每公斤體重l毫克,分3-4次肌肉注射。療程半年到3年。
分類
寄生蟲感染性侏儒症
1、日本血吸蟲
血吸蟲病在我國流行較為廣泛,危害嚴重,現已在一些地區得到控制。根據以往調查資料,一個地區血吸蟲病流行嚴重,居民感染率高,感染度重,血吸蟲病保儒症的發生率也高。
解放初期,黃銘新等(1956)在上海青浦縣錢盛鄉血吸蟲病流行區調查了三個村莊,共623人,當地居民血吸蟲病感染率為98%。在排除其他有關因素可能影響後,發現因息血吸蟲病而引起的侏儒症有25人,占總人口數的4%。在血吸蟲病嚴重流行的江西省玉山縣,一次收治住院的血吸蟲病人中,競有25%是侏儒症患者。1958年大規模收治血吸蟲病人時,各地報告的保儒症,在一般血吸蟲病患者中約占1.5%,在晚期血吸蟲病人中占5.8%一8.1%。血吸蟲病性侏儒症患者的男女比例約為2:1,年齡大多為16—20歲,個別為30歲,35歲以上的侏儒患者較少見,可能是多已病死的關係。
2、華支睪吸蟲
人因吃不熟的魚、蝦誤將該蟲囊蚴食入體內而感染。成蟲寄生在人的膽管內,可引起阻塞性黃疽、膽道炎;膽石症、肝腫大、肝實質萎縮和肝硬化等症狀,重症兒童患者可出現生長發育不良和侏儒症。華支睪吸蟲病在我國較為常見,呈散在流行。據調查,感染1000條成蟲的病例並不少見。朱師晦等(1981)在廣東查治了2214名本病患者,發現其中侏儒症患者32人,由華支睪吸蟲感染引起的侏儒症為1.44%。河南省衛生防疫站(1974)在該省淮陽縣調查了108名華支睪吸蟲病人,其中有生長發育障礙者7人,占6.48%。1976年,他們又在沈邱縣收治137名本病患者,其中有生長發育障礙者23人,占16.8%,比例頗高。山東省寄生蟲病防治研究所(1975)在東阿縣發現93例15歲以下的本病患者,21例有明顯的生長發育不良;在臨沂縣的一次調查中,發現92例華支睪吸蟲病人,3例出現嚴重生長發育障礙,其中侯窩大隊患者侯××,男,26歲,身高僅120cm,為典型的侏儒症病人。
3、鉤蟲、瘧原蟲、薑片蟲、黑熱病原蟲、鏇盤尾線蟲等
據文獻記載,這些寄生蟲的重度感染也會引起兒童的生長發育受阻。少數出現侏儒症。
垂體性侏儒症
垂體性侏儒症系由於垂體前葉功能不足所引起的生長發育障礙
病理變化
以垂體萎縮為主,垂體前葉分泌生長激素不足以致發育遲緩,也可能由於下丘腦生長激素釋放因子缺乏。生長激素直接作用於全身的組織細胞,可以增加細胞的體積和數量,促進機體生長。生長激素主要為一種蛋白質合成激素,它能加速胺基酸由細胞外轉運到細胞內,刺激DNA的複製及RNA的轉錄,生長激素能減少外周組織對葡萄糖的利用,血糖因而升高,它能抑制葡萄糖向細胞內轉移,與胰島素促進葡萄糖向細胞內運轉的作用恰好相反,因此,它與胰島素在糖代謝的調節中存在著相互拮抗的作用;此外,它能促進脂肪的動員與利用以供應能量。當生長激素缺乏時則引起生長緩慢甚至停滯。
多數患者同時有垂體促性腺激素分泌不足,部分病例也有促甲狀腺素、促腎上腺皮質激素分泌不足而引起有關內分泌腺的功能障礙。
臨床表現
原發性垂體性侏儒症多見於男孩,初生時身長體重往往正常,最初,一、二年與正常小兒差別不明顯,自1~2歲以後開始生長速度減慢,停滯於幼兒期身材,年齡越大落後越明顯,至成年其身長也多不超過130厘米,但智力發育正常,患兒外觀比較其實際年齡為小,但身體上部量與下部量的比例常與其實際年齡相仿,故各部分發育的比例仍相稱。患者頭稍大而圓,毛髮少而質軟,皮膚細而滑膩,面容常比其實際年齡幼稚,胸較窄,手足亦較小。(見圖)出牙延遲,骨化中心發育遲緩,骨齡幼稚與其同身高年齡小兒相仿,骺部融合較晚。
多數患兒性腺發育不全,第二性徵缺乏,至青春期男性生殖器仍小如幼童者,隱睪症頗常見,聲調如童音。女性往往有原發性閉經,乳房,臀部均不發達,身材無女性成年人特徵,子宮小,外陰如小女孩。甲狀腺、腎上腺皮質功能亦往往偏低,但臨床症狀常不明顯。
繼發性垂體性侏儒症可發生於任何年齡,得病後生長發育開始減慢並伴有原發病的症狀,患顱內腫瘤者可見顱內壓增高和視神經受壓迫的症狀及體徵,如頭痛、嘔吐,視野缺損或縮小等,甚至由於垂體後葉或下丘腦也受損害而並發尿崩症。
診斷
主要根據病史及體檢診斷。1~2歲以後生長緩慢,身長低於同年齡正常小兒三個標準差(S.D.),身體各部分發育比例相稱,智力正常。尚應注意檢查是否有原發疾病。
近年來血清中生長激素的濃度已能測定,患者常明顯降低。正常人休息狀態下空腹(早餐前)血清中生長激素濃度為3毫微克/毫升,兒童為5毫微克/毫升,患兒常低於5毫微克/毫升。由於其量甚微,測定不易準確。正常人於沉睡時或於進餐、運動以後血清生長激素較原來增加,故可於入睡60~90分鐘後取血或於進餐後2小時再運動10分鐘後取血,患者往往不增加或<5毫微克/毫升,有人套用胰島素或L-精氨酸靜脈滴注以產生低血糖,在正常人可促使血清生長激素有暫時性升高,而患者卻無反應。
垂體性侏儒症患者往往同時有垂體促甲狀腺激素及促腎上腺皮質激素分泌不足,其血清膽固醇可增高,血清T3及T4往往降低或在邊緣水平。尿內17羥類固醇與17酮類固醇排量都減低。
鑑別診斷
對身材矮小的小兒應注意與以下情況相鑑別:
一、慢性疾病所引起的生長發育障礙如慢性感染、慢性肝病、營養不良、先天性心臟病、先天性腎小管疾病。
二、其它內分泌代謝疾病如呆小病、卵巢發育不全,軟骨營養不良,糖原累積症及粘多糖增多症等。
三、家族性矮小父母身材矮小或家族中有矮小者,但智力正常,性發育亦正常。
四、青春期延遲症(體質性生長延遲症)患兒青春期延遲,不但性發育較遲,其體格發育包括骨骼發育比正常小兒可落後2~4年,智力正常。一旦到達青春期後即有很大變化,生長速度加快,最後能達到完全正常的高度。
五、原基性侏儒症病因尚不清楚,患兒生長激素正常,從胎兒期開始發育延遲,因此出生時體格即很細小,嬰兒期即顯侏儒。智力正常。有些病例尚伴有其它畸形,如頭、眼,耳、頸及心臟的異常變化。
治療
套用人類生長激素替代補充療法,可有80%患者有效,劑量每次1~2毫克肌肉注射,每周2~3次。一般第一年療效顯著,2~3年後稍差,可能由於對生長激素產生抗體之故。動物生長激素療效不佳且易產生抗體。人及猴的生長激素來源不多,一個垂體只能提煉5毫克生長激素,目前尚不能人工合成,也不能廣泛應。
同化激素可增強蛋白質合成促進生長,目前多採用苯丙酸諾龍,但此類藥物也可促進骨骺的融合而縮短生長時期,最後反而使身體形成矮胖,特別在用大劑量時更易如此,因此必須慎重。一般在10~12歲開始套用,每月每公斤體重1~2毫克,分為四次每周肌肉注射一次。用藥前應先檢查骨齡,每三月隨訪一次,一旦骨齡巳接近實際年齡即應停藥。
絨毛膜促性腺激素每次500~1000單位,每周二次肌肉注射,4~6周為一療程,對性腺及第二性徵的發育有刺激作用,男性患者療效較好。
此外尚可套用甲狀腺片,從小劑量開始,一般每天20~40毫克,對甲狀腺功能偏低的患者更有意義。有低血糖及腎上腺皮質功能不足的患者可予生理劑量氫化考地松(15~25毫克/日)或強的松(2.5~5毫克/日)口服。
對繼發性垂體性侏儒症患者,如顱內腫瘤可進行手術治療,其它原發疾病可給以相應的治療。
中醫辨證
中醫病名:虛勞,重子癆
定義及釋義
垂體性侏儒症系指機體青春期以前腺腦垂體功能減退,生長激素(growth hormone,GH)分泌缺乏或不足,或對生長素不敏感所致的生長發育障礙。
病因
中醫病因:本病病因可歸咎於先天因素和後天因素兩個方面。先天因素多由於父母精血虧虛而影響胎兒的生長發育;後天因素或因食物中毒,或因罹患溫熱疾病,或為高熱灼傷津液,耗傷氣血、精髓,致使脈絡失養,影響生長發育;或因久病致脾腎虧虛,氣血不足,不能滋養腦髓所致。病因為腦瘤者多由痰濁化毒凝結所致,但痰毒凝結成瘤,或痰瘀凝結成瘤,又與機體陰陽失調,臟腑失和有關,若由於創傷、手術、放射治療等因素起病者,均有正氣虛虧。
西醫病因:本病可分原發性和繼發性兩類,原發性者病因未明,多為先天性發育不全或遺傳疾病所致,可能為常染色體隱性遺傳,呈家族性,以單獨生長激素不足為主,男孩較女孩多見,男女比例約為2~4:10。繼發性者可繼發於下丘腦-垂體疾病,如腫瘤、感染、顱腦外傷、手術或放療等因素,直接損傷垂體,或損害下丘腦、或使垂體門脈系中斷而致病。下丘腦一垂體部位腫瘤為繼發性垂體性侏儒症的重要原因。
病機探微:中醫認為本病的發生與脾腎虧損,氣血不足,水濕內聚,陰陽俱虛有關。腎為先天之本,藏精,主骨、生髓、通於腦;腎主生長、發育、生殖,腎精虧虛,則生長、發育停滯,加以先天不足,致脾胃受損,氣血虧虛,肝失所養,筋骨痿軟,以致身材矮小,發為侏儒。
病理生理學:原發性垂體侏儒症的垂體變化有:先天性垂體缺如,先天性垂體發育不全伴嗜酸細胞缺乏或減少。垂體萎縮、纖維化或囊性變伴鈣化。除垂體改變外,常伴有性腺、甲狀腺和腎上腺皮質不同程度的萎縮,內臟及骨骼生長停滯於幼年階段。繼發性者以下丘腦一垂體部位腫瘤較為多見,其中以顱咽管瘤最多,垂體嫌色細胞瘤較少發生於兒童,但也可起病甚早而造成侏儒症。
案例
最著名的侏儒
拇指湯姆將軍(即查爾斯·S·史崔頓)是1838年由身高正常的父母所生下的孩子。湯姆11歲在P.T.巴納姆帶他進入馬戲團並以“來自英格蘭的將軍”的名義在海報上宣傳時,他身高只有25英吋,體重只有15磅。拇指湯姆成了一個名人並游遍全世界,會見領袖和皇室人員,包括亞伯拉罕·林肯、英國女王維多利亞以及王子阿爾波特。拇指湯姆結婚時,娶的是矮小的新娘利維尼厄·沃倫,兩人站在一架大鋼琴上招待兩千名來賓。拇指湯姆於1883年7月15日在他45歲時死於中風。一萬多人參加了他的葬禮。
最小的小人
露西婭·拉薩特是有史以來最小的女人。她1863年出生時只有8盎司重,7英寸高。她長大成人時,身高也只有20英寸,體重不超過8磅。當她穿著維多利亞風格的褶邊裙、坐在一個大人的大腿上時,她很容易被誤認為是一個陶瓷娃娃。她是當時身價最高的穿插節目的魅力人物,在月工資平均為20美元的時候她每小時掙20美元。儘管有對她脆弱的生命的專門護理,1890年,在乘坐一列火車旅行時,她還是不幸去世了。當時,火車因暴風雪停在了落基山上。車上沒有暖氣,不管她身上蓋上多少層被子,她弱小的身體都不能保持溫暖。她像一座玩具塑像僵在那兒,最終被凍死了。
小矮人島
2004年,在發掘印度尼西亞生存著巨蜥和小象的弗洛里斯島島上的一度繁盛的小矮人文明遺蹟時,考古學家有一個驚人的發現。距今九萬五千年到一萬兩千年的人骨表明了一個人的身高不超過三英尺的複雜社會的存在。科學家們還不確定在我們其他的、個子更高的祖先們統治著地球上其他部分時,他們為什麼能在與世隔絕的極樂世界孤立地存在那么長的年代。他們滅絕的原因看起來倒是確定的:島上的火山恰好於一萬兩千年前暴發過。
治療
骨細胞增高療法
“骨細胞生物激活增高療法”是中國人民解放軍鄭州防空兵學院醫院增高研究基地與北京生長發育研究中心專家組成特別科研攻關小組,聯合美國哈佛大學生物工程研究院。根據中國人的生活習慣、遺傳基因、生長發育等特點,並借鑑廣州、上海、武漢等地的增高長高技術,經過長達12年的盡心研究而研發出來的增高長高療法。“骨細胞生物激活增高療法”的成功問世,在世界衛生組織在日內瓦召開的全球人體骨骼研討大會上,獲得全球科技新成果金獎,得到了全球骨骼增高長高專家的一致認可,被指定為全球增高推廣技術。到目前為止,已經為100多萬例36歲以下的朋友解決了身材矮小的難題。
經絡離子介入療法
中國人民解放軍鄭州防空兵學院醫院增高研究基地在管理軍醫隊伍專業化,人員層次專業化,治療技術專業化,診療設備先進化等方面狠抓落實,率先在國內建立生長發育專科。匯聚了大批相關專業的專家教授,系統全面地開展了關於增高長高,發育遲緩,矮小症的臨床治療和研究,並取得了豐碩的成果。經絡離子介入療法是依託神經經絡--腦垂體--腎經--骨骼生長軸線,集多種中醫藥療法、多種效應於一體的複合性治療方法,具有見效快、療效佳的優勢。
罕見病詞條庫

生理學常見疾病
| 人體主要體統包括:心血管、呼吸、泌尿、生殖、神經系統、內分泌系統,認識疾病因包括以下幾個方面:疾病的病因、發病機制和病理改變,以及各系統疾病在發展嚴重時可能出現的共性病理過程。 |
內分泌系統及其疾病
| 內分泌系統(endocrine system)是機體的重要調節系統,它與神經系統相輔相成,共同調節機體的生長發育和各種代謝,維持內環境的穩定,並影響行為和控制生殖等。 |
