
疾病簡介
 嚴重複合型免疫缺乏症
嚴重複合型免疫缺乏症目前美國已有數州利用新生兒篩檢全面檢驗新生兒是否患有T細胞淋巴球數目低下,再去瞭解數目低下的原因,包括乳靡胸,迪喬西症候群患者,以及嚴重複合型免疫缺乏症患者。此方法現已成功診斷出一名嚴重複合型免疫缺乏症患者並已給予臍帶血幹細胞移植治療。
篩檢的敏感度並非100%,檢查結果會因為某些疾病有晚發作型、非典型、蛋白質攝取不足或是特殊飲食而有偽陰性的可能,如有健康上的問題,請諮詢相關的小兒科醫師。
症狀
嬰兒多在3-6個月大時出現症狀,主要表現為:1.生長遲緩。2.嚴重重複性的細菌、病毒、黴菌感染,以呼吸道感染、慢性中耳炎、鼻竇炎、皮膚感染、口腔黏膜感染等最常見,嚴重會導致全身性黴菌感染。3.皮膚黏膜嚴重損害會導致剝落性皮膚炎、皮膚紅疹、舌黏膜的深部潰瘍。4.長期慢性腹瀉,嚴重時可引起營養不良,甚至死亡。5.在台灣地區有感染非典型結核菌或卡氏肺囊蟲的案例。6.自身免疫疾病、過敏性疾病、血液系統異常或淋巴系統腫瘤。成因
嚴重複合型免疫缺乏症(severecombinedimmunodeficiency)泛指一群罕見先天遺傳疾病,患者因為缺乏體液及細胞免疫功能而導致嚴重重複性的感染,未加以治療的話,多數會在出生後一年內死亡。患者易感染包括細菌,病毒及黴菌,亦可以慢性腹瀉或生長不良來表現。發生率目前不明。以台大醫院為例,在1984~2001年間共有4例個案。以往嚴重複合型免疫缺乏症可依照免疫缺陷細胞的種類,而有T-,B+,NK-SCID,T-,B+,NK-SCID,andT-,B-,NK-SCID。其中以T-,B+,NK-SCID最常見。此T-,B+,NK-SCID又可依遺傳模式分為X-linked與autosomalrecessive兩種。隨著分子診斷技術的進步及吾人對免疫細胞發展與功能的逐漸了解,目前傾向以分子缺陷基因來命名,以避免傳統分類法對疾病診斷與遺傳模式的誤判。
診斷
最常見的SCID以X-linkedSCID為主。通常患嬰為男性,多在3-6個月大時求診,主要表現為生長不良,口腔及其他黏膜嚴重黴菌感染甚至導致全身性黴菌感染,反覆嚴重感染尤其以伺機性感染為特徵,在台灣地區的案例,也有數個個案是因為非典型結核菌株感染或肺囊蟲肺炎(pneumocystiscariniipneumonia)。理學檢查呈現生長不良,腹瀉,皮膚紅疹,沒有扁桃腺及淋巴結,但有時可見肝脾腫大。頭部X光通常看不到腺狀體,胸部X光通常看不到胸腺陰影。感染通常難以控制治癒,多在一歲前死亡。家族史中可能會有其他男性家屬於一歲前死亡的類似情形。初步診斷依靠家族史,臨床症狀,淋巴球分類及其他淋巴球功能測試。以X-linkedSCID為例,淋巴球總數通常較相同年齡者為低,淋巴球分類則證實沒有T細胞和自然殺手(Naturalkiller,NK)細胞,但仍有B細胞。這些B細胞卻無法產生足夠的抗體對抗病原菌,血清中IgA和IgM濃度偏低,IgG因為剛出生有母親的抗體因此數量正常,但會隨著年齡增長而減低,因此被歸類為無功能的B細胞。
確切診斷需依靠分子診斷,目前可做突變基因分析,分析患者基因的突變所在,進一步可提供帶因者基因檢測及產前診斷,以提供遺傳諮詢所需。
症狀治療
1.由於免疫力不全因此需避免接種活性疫苗,其中卡介苗,口服小兒麻痹疫苗最需注意。2.易有全身散發性感染,包括細菌,病毒和黴菌,因此需及時使用適當的抗生素,抗病毒藥物和抗黴菌藥物,並需給予預防性抗生素使用。
3.需定期補充免疫球蛋白以增強免疫力。
4.患者若需輸血,需使用沒有巨細胞病毒(CMV)並經放射線照射過後(irradiated)的血液製品,以避免巨細胞病毒感染或預防移植對宿主反應(graftversushostreaction)。
骨髓移植或臍帶血幹細胞移植
患者免疫力重建需依靠骨髓或臍帶血幹細胞移植。最好使用親屬間HLA(人類白血球抗原)相合的幹細胞移植。若為非親屬間HLA相合的幹細胞移植或親屬間HLA不相合的幹細胞移植則須特別注意術前準備工作及術後排斥graftversushostdisease(GVHD)的問題。術後仍須定期追蹤淋巴球數目,分類及功能。
酵素補充療法
若為ADA(Adenosinedeaminase)缺乏的病患,可考慮使用ADA酵素補充療法。
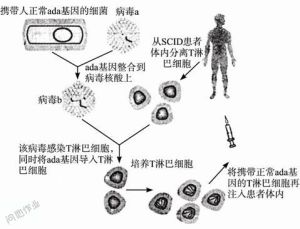
基因治療
由於骨髓移植的成功,而且HLA相合的捐獻者可遇不可求,基因治療可能是最根本的治療方法。這是以各種載體(vector)在體外矯正血球幹細胞的基因缺陷後,再將此改造後的幹細胞移植到病患體中。目前已有約二十名X-linkedSCID患者接受此種治療。初步成果與親屬間HLA相合的骨髓幹細胞移植相似,但至今已有3例個案出現嚴重副作用,因此此療法的安全性仍有待改進。
預後
若能早期診斷,並及早給予骨髓移植,目前成果顯示75~90%以上的嬰兒可以存活,親屬間HLA相合的骨髓幹細胞移植存活率最好。若使用其他的捐贈者的幹細胞,在移植成功後有部分仍需定期補充免疫球蛋白以矯正B細胞功能的缺陷。

