
基本概述
 高雪氏症
高雪氏症由於不同組織中酶活性不同可有不同臨床類型。本病為常染色體隱性遺傳,以猶太人中多見,約每50人就有1人攜帶異常雜合子基因,故發病率較高,可達8.3/10萬。中國已有不少報告。1982年楊氏綜合報導46例,北京兒童醫院30年共收治40例。
診斷
戈謝病的常見症狀為不明原因的脾腫大、肝腫大、貧血、血小板減少、骨痛、神經系統症狀等。診斷需結合臨床症狀、實驗室檢測及病理學檢查等進行綜合判斷。
葡萄糖腦苷脂酶活性檢測
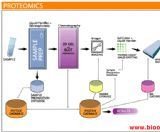
葡萄糖腦苷脂酶活性檢測是戈謝病診斷的金標準。當其外周血白細胞或皮膚成纖維細胞中葡萄糖腦苷脂酶活性明顯降低至正常值低限的 30%以下時,即可確診戈謝病。國內相關研究表明戈謝病患者的酶活性常低於正常值的 28%,不同實驗室由於檢測方法及參考值存在差異,酶學的檢測的結果可能有所不同,應該根據各實驗室的實際情況確定酶活性正常值。 但是值得注意的是,少數患者雖然具有戈謝病臨床表現,但其葡萄糖腦苷脂酶活性低於正常值低限但又高於正常低限 30%時,需參考該患者的血中生物學標記物(殼三糖酶活性等),並進一步做基因突變檢測,從而實現確診。
外周血白細胞葡萄糖腦苷脂酶活性檢測需採集新鮮全血樣本,並在短時間內分離白細胞。部分實驗室使用乾血紙片法( 或稱乾血斑法 Dried Blood Spots,DBS) 採集、運輸、儲存樣本,用於包括戈謝病在內多種溶酶體貯積症的診斷。該方法採樣方法簡便易行,僅需將患者新鮮全血滴在濾紙上,獲得直徑約 1cm 血斑,於室溫放置 4h 直至乾燥,然後將濾紙置於密封塑膠袋中運輸到中心實驗室,每個乾血斑約需 50μl 全血。乾血紙片法適合在遠離檢測中心的地區開展戈謝病酶學檢測的高危篩查,也適用於戈謝病的新生兒的篩查。
血漿殼三糖酶活性檢測可用於戈謝病患者的輔助診斷和治療效果的檢測。殼三糖酶是由活化的巨噬細胞在特殊環境下產生的,該酶的活性是目前戈謝病眾多生化標記物中升高最顯著的,患者的結果通常較正常人增高數百或上千倍。在套用酶替代治療後,治療有效患者的殼三糖酶活性會顯著下降,是能夠輔助診斷戈謝病及監測治療效果的生物學標記物。
骨髓形態學檢查
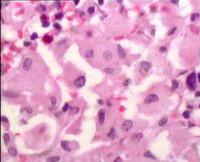
大多數戈謝病患者骨髓形態學檢查能發現特徵性細胞即“戈謝細胞”,該細胞體積,細胞核小,部份胞質可見空泡。但該檢查存在假陰性及假陽性的情況。 假陰性: 即當未查見戈謝細胞時,仍不能否定患有戈謝病,需要通過葡萄糖腦苷脂酶活性檢測進行確診。假陽性:骨髓中的單核巨噬細胞等會吞噬細胞碎片或脂質代謝產物,形成與“戈謝細胞”相似的“類戈謝細胞”,在慢性髓性白血病、地中海貧血、多發性骨髓瘤、霍奇金淋巴瘤、漿細胞樣淋巴瘤中都可能出現這種“類戈謝細胞”。因此,當骨髓中查見“戈謝細胞”時, 應高度懷疑戈謝病,但並不能確診戈謝病,需在鑑別區分其他疾病的同時,進一步做葡萄糖腦苷脂酶活性測定。
基因檢測
已發現的葡萄糖腦苷脂酶基因突變類型有 400 多種,相似的表型可有多種不同基因型,而相同基因型的患者,其臨床表現、病程及治療效果也不同。基因診斷並不能代替酶活性測定的生化診斷方法,但可作為診斷的補充依據並明確對雜合子的診斷。如果已通過酶學檢測確診戈謝病,可進行基因分子檢測,以預測患慢性神經性戈謝病的風險,以及確定合理的治療、隨訪方案。
診斷流程
由於戈謝病誤診、漏診率較高, 對於脾腫大和(或)血小板減少的患者, 可通過以下流程(通過文獻改編), 排除惡性腫瘤等疾病後, 進行葡萄糖腦苷脂酶活性檢測以確診或排除戈謝病。 本診斷流程圖從脾腫大開始,因為脾腫大是戈謝病最主要的特徵(在 ICGG 戈謝病登記研究有 87%的患者脾腫大為正常值的 5 倍),但是並不是所有的戈謝病患者都伴隨脾腫大,需進行綜合考慮。骨髓塗片細胞學檢出或未檢出戈謝病細胞都需要通過酶活性測定以確診。
鑑別診斷
白血病、淋巴瘤、多發性骨髓瘤、尼曼匹克病、地中海貧血等疾病臨床表現與戈謝病相似,需注意鑑別診斷。
治療
非特異性治療
可根據患者的臨床症狀與特徵選擇。貧血患者可補充維生素及鐵劑,預防繼發感染,必要時輸注紅細胞及血小板以糾正貧血或血小板減少。 骨骼病變的處理包括止痛、理療、處理骨折、人工關節置換等,並可輔以鈣劑及雙磷酸鹽治療骨質疏鬆。在無法接受酶替代治療的情況下,因病情進展(如脾功能亢進、脾梗死等) 可謹慎考慮脾切除,但應明確脾切除會加速葡萄糖腦苷脂在骨髓、肝臟、肺臟等器官的蓄積, 加劇許多臨床症狀、並增加多種致命的戈謝病併發症(如骨梗死、肺動脈高壓等) 的發生風險。脾腫大可通過觸診及影像學檢查( CT、 MRI)確定,脾切除的指征應結合患者的實際情況, 與外科醫生協作進行。脾切除的目的在於減小並維持脾的體積(≤正常值的 2~8 倍),減輕由於脾腫大帶來的症狀。脾切除後需要對肝、骨骼、肺的不良反應情況進行定期監測。
特異性治療
美國食品藥品監督管理局於 1991 年批准上市了由胎盤中提取的葡萄糖腦苷脂酶,後於 1994 年又批准了以基因重組方法研製的葡萄糖腦苷脂酶[注射用伊米苷酶(Imiglucerase)],用於戈謝病的 ERT治療。臨床實踐顯示,伊米苷酶可明顯改善Ⅰ型戈謝病患者的臨床症狀體徵,維持正常生長發育,提高生活質量,為Ⅰ型戈謝病治療的標準方法,治療越早,療效越好。伊米苷酶是截至2015年4月國內惟一可獲得的戈謝病特異性治療藥物。
治療進展
近年來, 美國 FDA 已經批准了一些底物減少療法,但該治療方法僅適用於成人,不適用於兒童患者, 且在中國尚無該類藥物獲準上市。分子伴侶療法、基因治療等正在探索中,臨床套用極少,尚無確切的證據證明上述治療方法對戈謝病的治療效果。
其他治療
對於兒童戈謝病患者,國外在上個世紀較為集中地展開過骨髓幹細胞移植治療,取得了一定成效。但是骨髓幹細胞移植存在死亡率高、異體移植匹配程度低等缺陷,並且目前尚未有隨機對照臨床試驗比較其相對於酶替代療法的有效性和安全性,國內亦無骨髓幹細胞移植成功治療戈謝病的病例報導。因此,骨髓幹細胞移植不應在能夠接受酶替代療法的患者中開展。
疾病管理
疾病管理計畫
戈謝病臨床表現個體間差異很大,其治療需要結合臨床表現、疾病負擔及生活質量的需求進行個體化。自確診戈謝病開始,需要對患者進行治療前臨床評估,設定治療目標,根據患者的臨床風險確定個體化 ERT 治療劑量,並進行疾病定期監測。 必要時對 ERT 治療劑量進行調整,以維持合理的治療目標。 在確定治療目標的同時,應該兼顧患者的經濟狀況, 相關的支持治療及姑息治療也可以考慮。
治療前臨床評估
在經葡萄糖腦苷脂酶活性檢測確診戈謝病後,需要對患者進行全面、可重複的治療前臨床評估[13, 25],為評估疾病風險、確定個體化治療劑量和治療目標、監測治療效果奠定基礎。
治療目標
伊米苷酶 ERT 為截止2015年4月 I 型戈謝病治療的標準方法。根據疾病風險評估確定合理劑量並規律治療 12~24 個月後,患者應達到以下治療目標。對於開始治療較晚、病情較重的患者,達到治療目標所需要的時間可能較長。
戈謝病 ERT 的治療目標:
骨骼:無骨危象,無骨痛或輕度骨痛;
血液學:血紅蛋白≥110 g/L(女性及兒童)或≥120 g/L( 男性),血小板計數≥100×10/L,無瘀斑及出血現象;
臟器:脾臟體積≤健康人 2~8 倍(根據梗死面積),改善食慾及腹痛,消除脾亢;肝臟體積≤健康人 1.5 倍,肝功能正常;
生活質量:在 2~3 年內改善生活質量評分。
持續臨床監測
持續監測疾病相關臨床參數變化是保證患者獲得個體化治療的關鍵。需根據患者是否接受 ERT 及是否達到治療目標,對患者進行定期檢查與評估。
遺傳諮詢和產前診斷
戈謝病是一種常染色體隱性遺傳病。患者父母如果再次生育,其子女患病的風險為 25%。對於生育過戈謝病患者的家庭及親屬應進行遺傳諮詢及致病基因攜帶者檢測。產前診斷是預防高危家庭再次生育類似患兒的最有效方法。在高危孕婦妊娠 9~13 周左右取胎盤絨毛,或是在妊娠 16~22 周左右取羊水進行胎兒羊水細胞培養,進行葡萄糖腦苷脂酶活性和(或)DNA 基因突變檢測。
發病機理
 高雪氏症
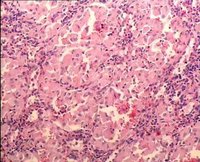
高雪氏症本病由於β葡萄苷酶(β-glucosidase)缺乏,致使葡萄糖腦苷脂(gluccerebroside)蓄積在肝、脾、骨骼和中樞神經系統的單核-巨噬細胞內,而造成肝脾腫大、骨骼受累和神經系統症狀。 葡萄糖腦苷脂是一種糖脂,溶解於水,是由長鏈的氨基乙醇神經鞘氨醇(aminoalcoholsphingvside)和長鏈的脂肪酸在C2的部位相連,此種化合物稱為N-醯基鞘氨醇(ceramide),一個分子的葡萄糖由β糖苷連結於鞘氨醇的C1部位而合成。正常情況下葡萄糖苷脂經β葡萄糖苷酶水解葡萄糖和N-醯基鞘氨醇。
由於β葡萄糖苷酶缺乏,則葡萄糖腦苷脂蓄積。在巨噬細胞內蓄積的腦苷脂來源於:①衰老紅細胞中的紅細胞糖苷脂(globoside),它是紅細胞糖脂中的主要成份;②衰老的白細胞和血小板中的主要糖脂-乳糖基醯鞘氨醇(ceramidelactoside);③血型物質中的鞘磷脂(Bloodgroupglycosphingolipids)。腦中堆積葡萄糖腦苷脂來源於神經節苷脂(Tanglioside);此外,由於神經鞘脂類是哺乳動物細胞膜的組成部分,因此蓄積的葡萄糖腦苷脂尚可來自體內各種組織如肝、腎和肌肉組織等。
正常人每克脾組織(濕重)含葡萄糖腦苷脂60~280μg,病人的含量為3~40.5mg,高出正常近百倍,但其他中性鞘氨醇糖脂類和半乳糖腦苷脂(galactocerebroside)則含量正常。
臨床表現
 高雪氏症
高雪氏症由於酶缺乏的程度不同,症狀可有較大差異;但同一家族中發病的都是相同的類型。根據各器官受累的程度發病的急緩,以及有無神經系統受累,分類在型:①成人型或慢性型;②嬰兒型或急性型;③少年型或亞急性型。 1.Ⅰ型(慢性型)起病緩,可見於任何年齡,以學齡兒童發病者最多,以往稱為成人型是不夠恰當的。此型最多見,楊氏統計的46例中,此型占15例。基β-葡萄糖苷酶的活力約相當於正常人的12~45%,發病早的具酶的活力相對地較低。
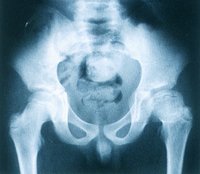
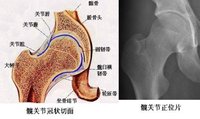
起病隱匿,病程緩慢,常以肝脾大和貧血就診。隨病情進展,可出現皮膚、眼和骨關節症狀,但無神經系統症狀。按病情進展可分三期:①初期:一般狀況好,僅有脾腫大和輕度正色素貧血,生長發育接近正常。②中期:肝臟亦逐漸增大,但不如脾大明顯。淺表淋巴結多不腫大。隨著貧血的加重,面色逐漸蒼白。由於脾功能亢進,白細胞和血小板亦多減少,網路紅細胞輕度增高。在暴露部位的皮膚呈現特殊的棕黃色。部分病人骨關節症狀出現較早,可有骨和關節隱痛。③晚期:各型症狀逐漸加重,貧血顯著,白細胞與血小板明顯減少,粒細胞甚至可低至10000/L以下,常合併感染和有皮膚黏膜出血傾向。淋巴結亦可輕度腫大。若肝臟浸潤嚴重,可出現肝功能損害,甚至肝硬化,食道靜脈曲張和凝血因子的減低,尤其Ⅸ因子缺乏比較多見。骨和骨髓浸潤可致骨痛,關節腫痛,有時需與風濕性關節炎鑑別。Ⅹ線檢查可見髓腔增寬、普遍性骨質疏鬆,並可見局限性骨質破壞;典型所見是股骨遠端膨大,有如燒瓶樣,常合併股骨頸和脊椎壓縮性骨折。化骨核癒合較晚。兩眼球結膜出現對稱性棕黃色楔形斑塊,其底在角膜邊緣,尖端指向眼眥,先見於鼻側,後見於顳側,此征多隻見於成年人,兒童較少見。此類患兒身高及體重多在正常低限。
2.Ⅱ型(急性型)發病多在1歲以內,可早在生後1~4周即出現症狀。較慢性型少見,楊金氏總結46例中此型占9例。此型β葡萄糖苷酶的活力最低,幾乎不能測出。
 高雪氏症
高雪氏症此型腦組織中葡萄糖腦苷脂的量尚不清楚。正常情況下,腦組織中的腦苷脂幾乎都是半乳糖腦苷脂。用薄層色譜測定,證實病人腦組織中尤其是額葉中蓄積的腦苷脂很大部分是葡萄糖腦苷脂。曾有報導此類病人灰質中的糖脂70%是葡萄糖腦苷脂,30%為半乳糖腦苷脂,而正常人腦灰質中,100%皆為半乳糖腦苷脂,說明高雪氏症Ⅱ型的腦組織中,至少是某些部位葡萄糖腦苷脂含量增高。
發病越早,病情進展越快。開始常出現消化不良症狀,以後則導致生長發育遲緩。除肝脾腫大和貧血外,主要是神經系統症狀,如意識障礙、斜視、頸強直、角弓反張、四肢肌張力增強以及下肢呈剪刀樣交叉、牙關緊閉、咽下因難、喉喘鳴,亦可出現驚厥。肺內大量高雪氏細胞浸潤,當病情嚴重時多有咳嗽,甚至出現呼吸困難和青紫。X線可見肺內浸潤性病變,骨骼改變不明顯。
3.Ⅲ型(亞急性型)可在嬰兒或兒童期發病,楊氏統計此型占9例。基β葡萄糖腦苷脂酶活力相當於正常人的13%~20%。作、斜視或水平注射注視困難和娃娃眼。腦電圖廣泛異常。病情進展時,四肢漸僵直,全身肌肉消耗萎縮,行走困難,語言障礙。北京兒童醫院曾見一家三個男孩,其中兩例雙胞胎同時發病,均於6歲行脾切除術,以後出現癲癇樣發作,第三個孩子以後也出現同樣的神經系統症狀,此型與Ⅱ型不同點除發病年齡外,一般無嚴重智力障礙,智商在70左右,以此與Ⅱ型鑑別。
晚期出現骨髓症狀,偶見病理骨折,由於血小板減少,常有出血症狀。
在楊氏分析的46例中有12例因診斷時年齡較小,尚未出現神經系統症狀,須追蹤觀察方可定型。
疾病預後
 高雪氏病:A.股骨密度減低,皮質變薄,遠端輕度擴張,呈“燒瓶樣”畸形。B.胸腰段椎體變扁楔變,脊椎後突畸形,骨質疏鬆。
高雪氏病:A.股骨密度減低,皮質變薄,遠端輕度擴張,呈“燒瓶樣”畸形。B.胸腰段椎體變扁楔變,脊椎後突畸形,骨質疏鬆。Ⅱ型多發於發病1年內死於繼發呼吸道感染,少數可存活2年以上。Ⅲ型在神經系統狀出現後,逐漸消耗,並有運動障礙,多死於反覆發作的繼發感染。Ⅰ型進展很慢,脾切除後可存活至正常人的年齡,智力完全正常。

