
概述
 基因圖譜
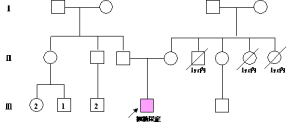
基因圖譜名稱:楓糖尿症(MSUD)
英文:maplesyrupurinedisease
基因定位:19q13.1;lq31......
楓糖尿症是常染色體隱性遺傳性代謝病,首先由Menkes等發現。各類型普遍都存在支鏈胺基酸降解酶的缺乏,並早期出現智慧型發育障礙和其他神經症狀。因尿中排出的代謝產物有類似"楓糖漿"味,而命名楓糖尿症發病率約1/100,000~1/300,000活產兒,中國自1987年起有報導。
流行病學
所有型MSUD均為常染色體隱性遺傳疾病。相關酶不同亞基上的缺陷可以解釋該病在不同家族中臨床和生化表現上豐富的多樣性。經典型患者可能存在E1α,E1β或E2亞基缺陷。E1α的編碼基因位於第19染色體上,E2基因位於第1染色體上。輕型和間歇型可能為2個不同等位基因突變的“雙雜合體”測定白細胞及纖維原細胞中的酶活性使診斷雜合體和染病胎兒成為可能發病率在美國大約為1/20萬;然而,在Mennonite教徒中經典型更為常見。
病理
病兒神經系統正常發育過程受阻,大腦皮質除海馬以外各部位層次不清,細胞結構也不成熟。神經母細胞的外向移動受阻,以致皮質以外有異位灰質存在(常見於腦室的室管膜周圍),說明在妊娠後期1/3中樞神經系統的發育有明顯受阻。其次,幾乎所有中樞神經的髓鞘化過程都受損,在大腦半球白質、錐體束、小腦齒狀核和胼胝體等部位尤其明顯,僅有內囊、視束、嗅束和脊髓後索不受影響。此外,灰白質中有廣泛的囊性改變,顯示空泡形成,周圍有多數星形膠質增生,形成海綿狀態。用組化方法染色發現空泡內含PAS陽性物質,但未發現髓鞘裂解的產物。
發病機制
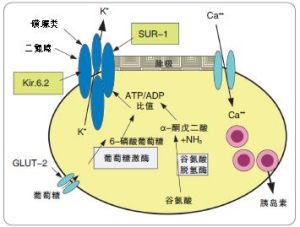 楓糖尿症
楓糖尿症臨床表現
大致可分四種主要類型,還有罕見的第五型:
1.經典型楓糖尿症
患兒在出生時狀況良好,一般從生後幾天或1-2個月內發現餵養困難,啼哭聲弱,不能吸乳和反應遲滯,以後即逐漸消瘦,智慧型低下,同時呼吸變淺,間斷出現發紺現象。體檢時可見全身肌張力減低或增高,Moro反射減弱或消失,強直性驚厥或角弓反張等都較常見。小兒囟門常膨出而緊張,還有時出現眼震、眼肌癱瘓、瞼下垂、瞳孔散大及對光無反應等。病情進展迅速,尿中逐漸出現特異氣味。本型在文獻報告的病例中約占70%,一般症狀較嚴重,預後較差,多死於酮中毒。
2.間歇性楓糖尿症
早期發育正常,反應也不遲鈍,大約從生後10個月到2歲間歇性出現厭食、嘔吐、表情淡漠、步態不穩、共濟失調、嗜睡和行為改變等。尿中有特異的氣味。病程長短不一,可以有多次起伏,也有的患兒發生急性酮中毒而死亡。本型約占20%。
3.輕型楓糖尿症
表現為精神發育遲滯,但無其他典型神經症狀和體徵,也沒有間歇發作的特點。
4.對VB1反應型楓糖尿症
僅有輕度智慧型發育遲滯,也無典型的或間歇神經損害症狀。僅有血中支鏈酮酸的含量比正常兒稍高。本型患兒對維生素B1的療效好,血中的生化異常可有顯著的好轉。
5.(E3)缺乏型楓糖尿症
極為罕見,臨床表現類似中間型,但由於E3亞單位的缺陷,患兒除支鏈α-酮酸脫氫酶活力低下外,其丙酮酸脫氫酶和α-酮戊二酸脫氫酶功能亦受損,故伴有嚴重乳酸酸中毒。患兒在出生數月內通常不出現症狀,隨著病程進展,逐漸出現進行性的神經系統症狀,如肌張力減低、運動障礙、發育遲滯等。尿液中大量排出乳酸、丙酮酸、α-酮戊二酸、α-羥基異戊酸和α-羥基酮戊二酸等;由於丙酮酸的大量累積,血中丙氨酸濃度亦增高。本型患兒限制蛋白和脂肪攝人,套用大劑量VB1等治療均無效。
檢查
用色譜分析法可測出經典型患兒血中幾乎所有的支鏈胺基酸含量都明顯增高(如亮氨酸平均增高30倍,異亮氨酸平均增高16.5倍,纈氨酸平均增高8倍),有時還在血中發現異亮氨酸的立體異構物別異亮氨酸(alloisoleucine),其他的胺基酸無大變化,同時還可測出血漿和尿中支鏈酮酸(NICA、KIVA、KMVA等)也大量增多,尿中a-羥基丁酸和a-羥基異戊酸等也常增高。在細胞培養中,可發現KICA和KMVA脫羧基酶活性顯著降低,KIVA脫羧基酶活性也可下降。此外,患兒還經常出現低血糖(空腹血糖有時低於600mg/L),葡萄糖耐量曲線呈低平型,可能是繼發於糖代謝障礙,血中胰島素過高或者糖原異生作用受損的關係。並有血中pH、pC02、HCOs含量下降,尿酸、尿中吲哚乙酸類排出增多等繼發的代謝紊亂。
診斷
對智慧型發育遲滯患兒的常規尿篩查中,如發現DNPH定性反應陽性,有可能是本病的早期患兒。對於疑似典型或間歇型的患兒,可根據其嚴重智慧型低下和尿中特異氣味來診斷。但確診必須有血或尿支鏈胺基酸的色譜分析,或用患兒的白細胞或皮膚成纖維細胞培養,直接測定支鏈胺基酸的脫羧基酶活性。基因攜帶者的檢出可使用細胞培養並測定脫羧基酶活性,患兒的父或母,凡是酶活性下降到正常兒的50以下者,就可能是基因攜帶者。妊娠期羊水細胞的培養,對10周以上胎兒測定KICA脫羧基酶的活性,有助於產前診斷。
防治
 治療的奶粉
治療的奶粉本病的治療原則是膳食控制。在患兒的食品中嚴格限制支鏈胺基酸類,蛋白總量也需限制在每日2g/kg體重以下,並需經常檢查血中的胺基酸濃度來參照。患兒在感染、外傷或有腸胃疾病時,由於代謝性失代償作用而致症狀加重。因此,此時必須採用鹼性溶液矯正酸中毒,減少蛋白人量和保持負氮平衡,必要時需反覆輸血或腹腔透析以挽救生命。此外,本病應常規套用維生素B1每日10~20mg,至少維持2~3年。對於產前診斷為陽性的胎兒,應儘可能地勸告其終止妊娠。
【附】其他有關的支鏈胺基酸代謝病
1.高纈氨酸血症
首先由和田(Wada)等(1965)報告的罕見代謝病。其代謝缺陷可能是纈氨酸轉氨基酶的活性缺乏。臨床表現與楓糖尿症相似,也有難餵養、嘔吐,不能吸乳等特點,但並不嗜睡,且常出現眼震,和楓糖尿症不同。患兒血和尿中纈氨酸增高,而其他胺基酸含量正常。通過限制支鏈胺基酸膳食可改善臨床症狀。
2.異戊酸血症
首先由田中(Tanaka)等(1966)報告的脂肪酸代謝障礙。病因可能是亮氨酸降解過程中異戊醯輔酶A脫氫酶的活性缺乏,以致其底物異戊酸在體內蓄積。臨床特點是患兒全身有特異的"乾乳酪"或"汗腳"的氣味,並表現有發作性酸中毒和昏迷,同時出現不同程度運動功能和智慧型發育遲滯,有些患兒從新生兒期就出現嘔吐和驚厥,並能迅速出現酸中毒而危及生命。實驗室檢查見血中異戊酸含量明顯增高(可達正常嬰兒的1500倍)。用C標記可見病兒末梢血中白細胞的1-14C-異戊酸的氧化明顯抑制。用亮氨酸內服作負荷試驗,也可見血中亮氨酸含量相應上升。治療方法同上。
3.B-羥基異戊酸(B_hydroxyisovaleric)和甲基丁烯醯甘氨酸尿症
這是另一種亮氨酸分解的代謝病,首先由Eldjarn等(1970)和Stokke等(1972)描述。代謝缺陷可能是甲基丙烯醯輔酶A羧化酶活性缺乏。臨床表現是出生2周左右出現上下肢肌張力低,運動發育遲滯。以後逐漸出現肌萎縮、腱反射缺失和舌肌纖顫等,與嬰兒型脊性肌萎縮(Werdnig-Hoffmann病)相似。患兒的尿常有特異的"貓尿"氣味,有時有助於診斷。在實驗室檢查中,尿液可測出大量的B_羥基異戊酸(HIV)和B_甲基丁烯醯甘氨酸(MCG)。從患兒白細胞或成纖維細胞培養中可測出酶活性。治療方面也以限制亮氨酸膳食為主(每日不超過150mg/kg體重),由於生物素是羧化酶輔酶,每日內服生物素250mg,可使患兒尿中排出的有機酸減少,但對臨床症狀有無療效尚未肯定。
4.甲基-p羥基丁酸尿症
同屬亮氨酸分解障礙,首先由Daum等(1971)報告。其代謝缺陷可能是硫解酶的活性缺乏,不能使亮氨酸的降解物a-甲基乙醯輔酶A轉化為丙醯輔酶A和乙醯輔酶A。患兒的臨床表現為反覆發作的代謝性酸中毒和智慧型發育遲滯,嚴重者伴意識障礙。尿中排出大量a-甲基-B羥基丁酸(MHBA),但無特異氣味。治療原則以限制蛋白攝入量(每日<2g/kg體重=為主 。

