
疾病概述
 遺傳性痙攣性截癱
遺傳性痙攣性截癱發病機制
 遺傳性痙攣性截癱
遺傳性痙攣性截癱L1CAM基因編碼的神經細胞粘附分子L1是粘附分子免疫球蛋白G超家族中的一員,主要在神經細胞中表達,與神經元一神經元粘附以及其他一些重要的神經元相互作用有關。Jouet等(1994),在HSP-1研究中發現了LICAM基因突變與HSP-1發病相關,突變形式可表現為錯義突變、無義突變及缺失突變。另外,LICAM基因突變還可引起X-連鎖的MASA綜合徵(MentalRetardation,AphasiaShufflinggait,AdductedThumbSyndrome)、X-連鎖的腦積水及X-連鎖的胼胝體發育不全。因此,我們稱這4種病為等位基因病(allelicdiseases)。由於這4種病的臨床特徵顯示有相當大的重疊,以胼胝體發育不全(corpuscallosumhypoplasia)、精神發育遲滯(mentalretardation)、拇指內收(Adductedthumbs)、遺傳性痙攣性截癱(hereditaryspasticparaplegia)和腦積水(hydrocephalus)為特徵,最近將這些疾病概括在一起,命名為CRASH綜合徵。
 遺傳性痙攣性截癱
遺傳性痙攣性截癱Hazan等(1999)研究發現Spastin基因突變引起HSP-4。Spastin是一種胺基酸ATP酶(AminoAcidATPase,AAA)蛋白家族的一個成員。HSP-4廣泛表達於人類成人及胎兒組織,定位於核中,與26S蛋白酶同源,可能與核蛋白生物功能和聚集有關。到目前為止,40%~50%HSP-4被發現有spastin基因的突變,約有39種,包括11種錯義突變、6種無義突變、10種剪接位點突變、8種小缺失突變、3種插入突變和1種大缺失突變等。
CiorgioLasari等(1998)在HSP-7病人中發現了Paraplegin基因的一種9.5kb缺失的雜合突變,另外他們還發現了兩種移碼突變,導致截短的Paraplegin蛋白的產生,確定paraplegin基因是HSP-7的疾病基因。Paraplegin是一種線粒體金屬蛋白酶,與酵母線粒體ATP酶高度同源,轉染的Cos-7細胞免疫螢光分析和體外線粒體表達實驗表明,Paraplegin蛋白存在於線粒體內膜,它有線粒體膜內蛋白水解作用,分子伴侶(chaperone)樣活性,線粒體蛋白翻譯後的裝配,多肽鏈的錯誤摺疊或翻譯等功能有關。在有Paraplegin突變的兩個病人的肌活檢分析中發現存在典型的線粒體氧化磷酸化缺陷,提示此缺陷可能是HSP-7型疾病神經變性的一種發病機制。
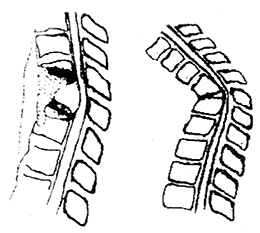
疾病病理
 遺傳性痙攣性截癱
遺傳性痙攣性截癱臨床表現
 遺傳性痙攣性截癱
遺傳性痙攣性截癱HSP分型:Harding(1984)的分型方法為大多數學者接受。Harding按臨床表現分為兩型:
1、單純型HSP,是臨床最常見的HSP。主要表現為痙攣性截癱,也有遺傳異質性,呈常染色體顯性遺傳,或常染色體隱性遺傳,病理改變主要在脊髓錐體束變性,而脊髓小腦束、後索改變不明顯。顯性遺傳的HSP又按年齡分為早髮型和晚髮型。早髮型最多見,常於35歲前發病,這型HSP患者行走較遲,雙下肢僵硬,不靈活,痙攣性癱瘓,腱反射亢進、膝踝陣攣陽性,病理征陽性。雙上肢可有輕微手指活動不靈活,腱反射活躍,深感覺障礙隨病程進展而越來越明顯。括約肌障礙和弓形足也可見。晚髮型患者常於40~65歲出現行走困難,臨床表現類似早髮型,但雙下肢肌無力、深感覺障礙、括約肌障礙更常見。
2、複雜型HSP,臨床上較少見,除痙攣性截癱表現外,常伴有脊髓病損外的伴發症狀體徵,遺傳異質性更明顯。
Ferguson-Critchley綜合徵:臨床特點是中年起病,四肢錐體束征,踝反射減弱或消失,其他腱反射亢進。四肢協調障礙,深感覺略減退。眼部症狀主要是眼球震顫,側向及垂直注視受限,假性眼肌麻痹。錐體外系損害表現四肢僵硬,不自主運動,面部表情少,可有前沖步態。呈常染色體顯性遺傳。
 遺傳性痙攣性截癱
遺傳性痙攣性截癱HSP伴早老性痴呆(Mast綜合徵):起病於20歲左右,痙攣性截癱,伴有構音障礙,痴呆,手足徐動症。呈常染色體隱性遺傳。痙攣性無力伴雙手和腿部小肌肉進行性萎縮、精神發育遲滯和中心性視網膜變性等;合併眼肌麻痹稱為Barnard-Scholz綜合徵。
HSP伴精神發育遲滯或痴呆:又稱魚鱗癬樣紅-痙攣樣截癱-精神發育遲滯(Siogren-Larsson)綜合徵:幼兒期發病或生後不久出現頸、腋窩、肘窩、下腹部及腹股溝等皮膚瀰漫性潮紅和增厚,隨後皮膚角化脫屑,呈暗紅色鱗癬,痙攣性截癱或四肢癱(下肢重),常伴假性球麻痹、癲癇大發作或小發作、手足徐動、輕至重度精神發育遲滯等;1/3的病例視網膜黃斑色素變性導致視力,可見視神經萎縮或審神經炎,但不失明;患兒身材矮小,牙釉質發育不全,指(趾)生長不整齊。預後不良,多在發病不久死亡,罕有存活至兒童期。呈常染色體隱性遺傳。
HSP伴遠端肌萎縮(Tyorer綜合徵):於兒童早期起病,主要表現為痙攣性截癱,假性球麻痹,伴有遠端肌萎縮、身材短小,輕度小腦症狀,手指徐動和耳聾等,部分病例不自主苦笑,構音障礙,到20~30歲不能走路。呈常染色體隱性遺傳。
HSP伴錐體外系體徵:如靜止震顫、帕金森樣肌強直、肌張力減低性舌運動和受阻徐動症等,最常見帕金森綜合徵樣痙攣無力和錐體束征。
HSP伴視神經萎縮(Behr綜合徵):通常合併小腦體徵也稱為視神經萎縮-共濟失調綜合徵,為常染色體隱性遺傳。10歲前逐漸出現視力下降,眼底視乳頭顳側蒼白,乳頭黃斑束萎縮,合併雙下肢痙攣、齶裂、言語不清、遠端肌萎縮、畸形足、共濟失調和腦積水等。完全型常於20歲前死亡,頓挫型壽命可正常,僅視力輕度下降。
HSP伴多發性神經病:表現感覺運動性多發性神經皮質脊髓束病變體徵,兒童或青少年期起病,至成年早期不能行走時病變才停止進展。腓腸神經活檢呈典型增生性多發性神經病。
Charlevoix-Sageunay綜合徵:多在幼兒發病,表現痙攣性截癱、共濟失調、智力低下、二尖瓣脫垂、雙手肌萎縮和尿失禁等。
遺傳形式
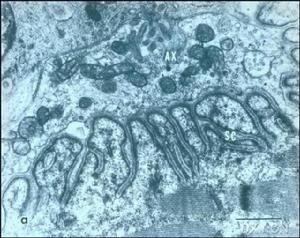 遺傳性痙攣性截癱
遺傳性痙攣性截癱遺傳性痙攣性截癱是一種比較少見的家族遺傳性變性病,最常見為常染色體顯性遺傳、也有常染色體隱性遺傳及x鏈鎖遺傳。以慢性進性無力與慢性痙攣性下肢癱瘓為特徵。發病機制至今仍不清楚。
鑑別診斷
 遺傳性痙攣性截癱
遺傳性痙攣性截癱HSP的診斷主要基於臨床症狀體徵,陽性家族史,並排除其他疾病。因此,HSP的鑑別診斷很重要,特別是對臨床特徵不典型及沒有相同疾病家族史的患者。目前,基因診斷已成為可能,但只限於已克隆的5型疾病基因的突變檢測。肌活檢有助於HSP-7型的診斷頸椎病常有上肢受累,神經根性疼痛,頸椎X線片及MRI示頸椎骨質增生。多發性硬化有緩解與復發的病史,視神經炎,MRI示腦部脫髓鞘改變。肌萎縮側索硬化有上肢肌萎縮,肌束震顫,肌電圖示巨大電位改變。Arnold-chiari畸形有共濟失調錶現,頭顱MRI可確診。脊髓小腦型共濟失調以共濟失調錶現為主,眼球運動障礙,構音障礙等。本病須與Arnold-Chiari畸形、頸椎病,多發性硬化、腦性癱瘓和遺傳運動神經元病等鑑別。
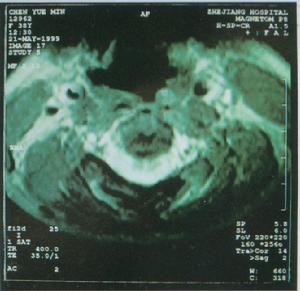
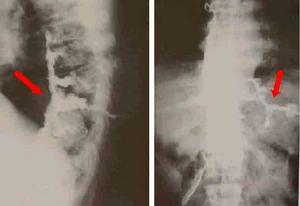
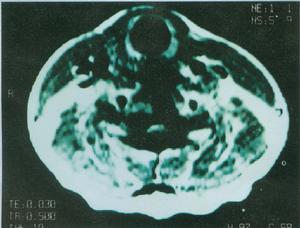
輔助檢查
 遺傳性痙攣性截癱
遺傳性痙攣性截癱2、肌電圖可發現視神經改變,但周圍神經傳導速度正常。
3、MRI頭顱MRI一般無異常,但某些病例可表現胼胝體發育不良,大腦、小腦萎縮。頸段或胸段脊髓MRI可顯示脊髓萎縮。
治療原則
 遺傳性痙攣性截癱
遺傳性痙攣性截癱2、針灸及康復治療。
3、早期可考慮基因治療。
用藥框限內的藥物只能緩解或減輕症狀,有條件時可選用營養藥物以增強體質、提高抵抗力。可肌注神經生長因子或細胞生長肽。
治療方法
不同的病情要選擇對症的遺傳性痙攣性截癱的治療方法。1、心理治療:針對心理不同階段的改變制定出心理治療計畫,可以進行個別和集體、家庭、行為等多種方法。
2、日常治療:主要是日常生活動作,職業性勞動動作,工藝勞動,使高位截癱患者出院後能適應個人生活、家庭生活、社會生活和勞動的需要。
3、主要是臨床康復:用特殊護理和苭物如維生素A、C/E,固齒強骨膠囊、鈣片等手段,預防各種合併症的發生,亦可進行一些治療性臨床處理,減輕症狀,促進功能恢復。
4、物理治療:主要是改善全身各個關節活動和殘存肌力增強訓練,以及平衡協調動作和體位交換及轉移動作,以及理療。
5、輔助器具:可以定做一些必要的支具來練習站立和步行,另外也可配備一些助行器等特殊工具,靠這些工具來補償功能的不足。
科研技術
神經修復學是中國骨與關節研究所一門獨立的學科,研究神經再生、神經結構修補或替代、神經重塑、神經調控。神經修復技術是中國骨與關節研究所研究的國際前沿的醫學技術,成功突破了脊髓損傷、截癱這個世界醫學難題。採用細胞移植等物理因素神經刺激或調控、藥物或化學等各種干預策略,在原有神經解剖和功能基礎上,促進被破壞或受損害神經再生修復和重塑、重建神經解剖投射通路和環路、調控和改善神經信號傳導、最終實現神經功能修復。這一技術誕生多年來成功地使數千名脊髓損傷截癱患者得到有效治療。治療預防
 遺傳性痙攣性截癱
遺傳性痙攣性截癱遺傳性痙攣性截癱,是一種遺傳病,沒有特效的治療方法,因此應將重點放在預防上。避免近親結婚,做好婚前檢查,本病患者儘量不結婚或結婚後不要生育,病程中應加強體育鍛鍊,防止過早臥床而致殘廢,本病發展緩慢,只要注意護理,可維持數十年生命。

