病理
 淋巴管平滑肌瘤
淋巴管平滑肌瘤肉眼見肺組織常為瀰漫的蜂窩狀結構,表現為大小不等的囊腫,直徑約1cm左右。
鏡下可見病變早期為未成熟平滑肌細胞堆積在末端支氣管外膜、肺泡壁和胸膜,肺泡毛細血管不累及。
病變後期,由於肌細胞聚積,膠原沉積增加形成結節及水腫、出血和巨嗜細胞減少。
破壞的肺泡可相互融合形成囊,囊壁襯覆扁平上皮和纖毛上皮,增生的結節可突進囊內、肺泡和細支氣管,使這些壁增厚及結構紊亂。
臨床特徵
 淋巴管平滑肌瘤
淋巴管平滑肌瘤2.反覆自發性氣胸,可表現為單側和雙側,有文獻報導,約95%的患者以自發性氣胸為首發症狀。
3.乾咳、較少見的有胸痛、胸腔乳糜滲出,痰中帶血和喘鳴。
4.肺外症狀有乳糜尿,乳糜心包積液,乳糜腹水。
5.其它:有些患者中可發現子宮纖維瘤或腎血管平滑肌脂肪瘤,肺功能改變:常為阻塞型通氣功能障礙和彌散功能障礙。
流行病學中國外尚無確切的發病率報告。該病自1937年首次記載至1997年,世界累計報導300餘例。美國丹佛間質性疾病研究中心10年僅報告16例,占瀰漫性肺間質疾病的比率<1%,因此LAM被認為是一種罕見病。中國有散在的病例報導。南京醫院鼓樓呼吸科1997~2000間經病理診斷了6例LAM,並認為該病可能並不罕見,過去診斷率低主要與對其臨床特點認識不足以及診斷手段不夠先進有關。非淋巴管平滑肌瘤幾乎均發生在絕經前婦女,70%發生在20~40歲之間,僅5%發生在50歲以後。極少病例發生在絕經之後,但這些病人多長期接受雌激素治療。
發病機制
LAM病因不明,許多事實提示本病可能與雌激素有一定關係。初潮之前不發生本病,絕經後也罕見,為數不多的發生絕經後的病例也常有補充雌激素素史。本病妊娠期加重,卵巢切除後後減輕,在表現為肺病變的結節性硬化症的病例中,女性占明顯優勢,活體組織也證實有雌激素和孕激素受體。但以抗雌激素治療,如切除雙側卵巢、放射治療、黃體酮等治療,收效並不理想,說明淋巴脈管平滑肌瘤病的發病機制除與雌激素有關外,還有其它重要因素參與。
至於肺間質平滑肌增生引起的肺囊腫形成及類似肺氣腫變化原因尚不清楚,有學者認為是由於平滑肌壓迫傳導氣道所致,但有爭議。另認為氣道內平滑肌增生形成“球-瓣”阻塞效應是引起終末氣腔擴張的主要原因。彈力蛋白/α1-抗胰蛋白酶系統的不平衡,導致彈力纖維的變性可能是引起肺囊腫和肺氣腫樣變化的主要原因。
病理
肺表面廣泛分布的囊腫是由擴張的淋巴管在肌層和壁層胸膜上形成類似肺大泡病變所致。新生的不典型平滑肌細
 淋巴管平滑肌瘤
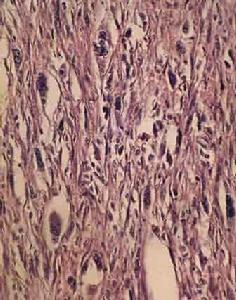
淋巴管平滑肌瘤上述異常增殖的平滑肌細胞其細胞學形態特殊,基因表形呈異質性。在LAM結節內可見三種不同的細胞型:較大的類上皮細胞位於結節外周,而較小的細胞居於中心。除形態學不同外,LAM細胞可套用抗人黑色素瘤抗體(HMB-45)和金屬蛋白酶(MMPs)染色顯示不同的免疫表型。在這些細胞記憶體在不同的蛋白顆粒:平滑肌特異性α-肌動蛋白、波形蛋白和結蛋白顯示不同的免疫組化染色反應提示平滑肌起源的異質性。在電鏡下,LAM細胞也與通常的平滑肌細胞不同。細胞核呈圓形或橢圓形、多切跡、核仁明顯、內含微絲狀物基質成分形成的緻密體和發育良好的內質網。由許多電子密度的膜結合顆粒構成細結晶狀層狀體。相似的細胞也見於腎、肝的血管平滑肌脂肪瘤和肺的透明細胞瘤、LAM細胞異質性使人們構想LAM的發病機制可能與血管或脈管相關的細胞類型似腫瘤樣異常分化與增殖有關。HMB-45是從黑色素瘤雜交瘤提純的一種單克隆抗體。病理學用於惡性黑色素瘤的診斷。在LAM細胞中呈陽性染色。HMB-45與兩種靶蛋白10kD的糖蛋白(Pmel17和GP100)結合。HMB-45陽性見於惡性黑色素瘤和其它細胞線來源的黑色素瘤,如LAM、血管平滑肌脂肪瘤和透明肺腫瘤以及結節性硬化症綜合徵(TSC),提示它可能來源於共同的前體產生的交叉反應。HBM-45曾用於組織學已知證實的5例LAM,均呈陽性結果,而15例原發性自發性氣胸、肺氣腫、Langerhan組織細胞增生症和IPF則均示陰性結果。
胸部影像學
(一)胸部X線片
 淋巴管平滑肌瘤
淋巴管平滑肌瘤淋巴管平滑肌瘤患者的胸片所見差異較大,早期可無明顯異常,或表現為磨玻璃影。隨病情發展漸出現瀰漫性小結節,從粟粒狀到中等大小的結節狀或網狀結節影,同時有不規則的網紋和線條狀陰影,多呈均勻性分布。這些陰影可能是多發性囊腔壓迫過度增生的平滑肌所致。早期肺容積正常。隨病情發展肺野中可見模糊不清的少量囊性變,一般囊腫直徑>1cm時胸片才能顯示。大量肺囊腫形成可使肺容積明顯增大,與肺氣腫相仿、淋巴阻塞可形成KeleyB線。同時可見單側或雙側胸液,常為乳糜性,量較多而且反覆發生。乳糜胸液也可發生在肺未受累的情況下,自發性氣胸發生率高,淋巴管造影可發現腹後壁病變。
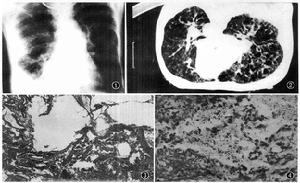
(二)胸部CT和HRCT
是診斷淋巴管平滑肌瘤的重要手段。胸部CT尤其是HRCT可以明確顯示出普通胸片顯示不清的肺囊腫。淋巴管平滑肌瘤的肺囊腫具有顯著特點,為全肺均勻分布的大小不等的薄壁囊腫,直徑在0.5~5cm之間,囊壁的厚度一般<2mm。早期囊腫較小,隨病情發展囊腫加大。這類形態的囊腫發生率為100%,是診斷淋巴管平滑肌瘤的重要依據。早期約50%患者出現磨玻璃影。在CT上能看到結節陰影者僅5%左右,為腫大的囊腫壓迫周圍過度生長的平滑肌細胞所致。如出現片狀陰影則提示出血。Sherrier等報告的8例LAM患者中4例發現縱隔淋巴結腫大。以HRCT定量測定囊腫面積與測定肺容量、彌散功能、運動試驗所評價的疾病嚴重程度相符合,故HRCT即可用於診斷又可判定預後。CT或HRCT可發現腹後壁、腹腔、腎臟、盆腔的病變。
肺功能檢查
LAM是間質性肺疾病中為數不多的胸片呈網狀結節影、肺容積增大而肺功能呈阻塞性或混合性通氣功能障礙的疾病之一。LAM肺功能表現為肺總量(TLC)增大,殘氣(RV)和RV/TLC增加,常見氣流受限,第一秒用力呼吸量(FEV1)和肺活量、FEV1/FVC下降。肺機構力學表現為平均彈性回縮力減低和上游阻力增加,彈性回縮力喪失和肺阻力增加均可引起氣流受限。
LAM常出現氣體交換異常,彌散功能(Dlco)顯著降低,PA-aO2增大。多數病人運動做功減低,耗氧量減少和無氧域降低,運動會導致呼吸頻率增快、每分通氣量增加和呼吸儲備減少。運動受限對運動能力也有顯著影響。由於氣流受限(通氣量下降)和肺血管功能失調或破壞,導致很多LAM病人運動能力的嚴重受損。
鑑別診斷
主要從臨床表現(育齡婦女,反覆發作氣胸史,LAM常伴有腹部改變)+囊狀影特徵(大小類似,囊外無結,囊內無動脈,囊間正常肺,壁薄,分布較均一)+少間質纖維化表現
 淋巴管平滑肌瘤
淋巴管平滑肌瘤(1)纖維肺泡炎和終末期間質纖維化(蜂窩肺):其囊性氣腔直徑為1mm~2.5mm,分布多不規則,壁較厚且伴有不規則小葉間隔增厚,此外尚有肺容積縮小,支擴和結構明顯變形,外周胸膜下改變明顯。
(2)神經纖維瘤病:此病亦可見囊狀氣腔,但其分布與LAM不同,囊氣腔位於肺尖部伴基底線紋理增強。
(3)支氣管擴張:其囊腔沿支氣管分布,且壁厚,常有液平,囊狀影在肺周圍少見,足以與LAM相鑑別。
(4)肺組織細胞增生症X:與LAM相似,但囊狀影的同時有多髮結節影,結節內見空洞,肺間質呈結節狀,網狀改變,且結節和囊樣病變多位於兩肺上葉,無乳糜胸水。
(5)肺氣腫:與LAM相仿,鑑別時較難。但肺氣腫缺乏小葉間隔增厚,多數囊狀低密度區中央可見殘留小葉結構,在低密度影之中可見小葉中央動脈;而LAM囊腔大小趨一致性,分布均勻,有明確均勻薄
壁,血管影位於囊狀影邊緣外,且一般無特大囊腔(類似肺大泡樣改變)。結合臨床病史,年齡,性別有助於鑒
別。
(6)結節性硬化症:是一種全身性疾病,當它侵犯肺時,與LAM在影像上病理上有共同之處。有文獻報導它可與LAM並存,亦有人認為LAM是結節性硬化症的一種頓挫型。兩者肺部表現臨床症狀均相似,但有作者認為系兩種不同的疾病,但從影像學上無法區別,結節性硬化症主要累及血管平滑肌而極少侵犯淋巴管和淋巴結,因此,乳糜胸罕見。此外本病有遺傳傾向,肺外多器管病變。
治療
本病尚無滿意的治療方法。糖皮質激素和細胞毒製劑無效。安宮黃體酮每月400mg或每2個月400mg肌肉注射,亦可選每日10~20mg口服,但療效並不理想。三苯氧胺和黃體酮激素釋放激素等也曾用於本病治療,但效果不能肯定。採用卵巢切除術聯合孕激素的治療方法較單一治療有肯定的療效。採用肺移植治療本病全世界已進行60餘例,50%患者可生存3年。移植肺也有再發LAM的報導。本病自然病程呈進行性進展,預後較差,中位數生存期為8~10年,多死於呼吸衰竭,偶有生存20年的病例。在病程晚期,偶可出現急性惡化,妊娠及雌激素可使疾病加重。肺功能和組織病理學可提示預後。肺總量增加和FEV1/FVC降低提示生存期短,組織病理學以囊性變為主比肌型改變為主者預後差。
預後
本病預後較差,大部分患者確診後1-2年即出現進行性肺功能障礙,最終死於呼吸衰竭。
淋巴管平滑肌瘤和組織細胞增生證X的影像學表現相似,前者多見於育齡婦女,常伴有乳糜胸。

