
 先天性脛骨假關節
先天性脛骨假關節疾病別名:
所屬部位:下肢
就診科室:骨科
症狀體徵:體型異常
病因
先天性脛骨假關節是由什麼原因引起的?
 先天性脛骨假關節
先天性脛骨假關節先天性脛骨假關節發病原因目前尚不完全了解,而學說頗多,如宮內損傷學說,血管畸形和代謝障礙學說等,但己被放棄,目前公認比較合理者有以下三種:
1、神經纖維瘤學說先天性脛骨假關節是由骨內性和骨外性的神經纖維瘤物質而引起,使局部骨質的正常發育障礙,骨折後也因此影響骨折的修復而造成假關節。Boyd報告14例先天性脛骨假關節的病例中,9例有皮膚的神經纖維瘤結節及皮膚色素斑。溫習了178例中有17例有此情況。還有個別病例在脛後神經有些變化。天津醫院所見9例先天性脛骨假關節與神經纖維瘤有許多相似之處,同時部分病例在假關節處的顯微鏡檢查也符合神經纖維瘤。這兩種病無疑有較為密切的關係。
2、纖維異樣增殖學說Boyd和Sage(1958)報告15例,其中兩例假關節處,顯微鏡下呈纖維異樣增殖的表現,提到與該病有關係。多數病例皮膚有咖啡色斑點,同時在假關節前期,囊性改變區的骨有彎曲畸形,甚至有些病例陰蒂較大,甚似纖維異樣增殖的第三型即Albright病。X線表現,在假關節是前期的囊性變區或假關節處殘留的囊性變區呈磨砂玻璃樣,骨小梁消失與纖維異樣增殖相似,更主要的是局部病理變化也頗為相似。從以上關係看來,二者之間亦存在更密切的關係。
3、神經學說Aegerter(1995)提出神經纖維瘤、纖維異樣增殖和先天性脛骨假關節三者有許多共同之處,可能由一共同的原因所引起的纖維組織錯構增殖。這些纖維組織的轉化不良和紊亂使局部區域產生骨質的正常生長和成熟顯得無能,這種變化的原因可能是局部神經通路不正常。在Moore的78例先天性畸形病例研究中有91%的病例周圍神經有病理改變,支持了這種神經學說。
症狀
先天性脛骨假關節有哪些表現及如何診斷?
典型的先天性脛骨假關節畸形是患兒的小腿中1/3處向前彎曲畸形或假關節外形,患肢短縮。
根據脛骨形態,臨床上一般分成三型。
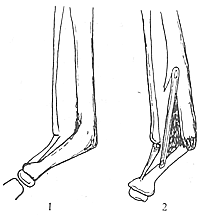
(一)彎曲型出生後脛骨下段向前彎曲,但無假關節,脛骨前弓處皮質增厚,髓腔閉塞,脛骨端萎縮硬化,呈前弓外形。發生骨折後,經一般處理局部不癒合,形成假關節。或因不認識此病,貿然作截骨手術,形成不癒合,繼續發展而兩斷端吸收,骨端硬化,遠端進一步萎縮變細,呈筆尖狀(圖1)。
 先天性脛骨假關節
先天性脛骨假關節圖1先天性脛骨假關節
(二)囊腫型出生後在脛骨中下1/3處呈囊性改變,但骨幹不細,臨床不易發現,輕微外力造成骨折後出現不癒合,繼之形成假關節。
(三)假關節型出生後即發現有脛骨中下段缺損,形成假關節。假關節處有較堅硬纖維組織連線或軟骨連線,骨端隨生長發育而變細,萎縮,遠端更為明顯呈筆尖狀,皮質菲薄。有時周圍軟組織也萎縮,包括腓腸肌。如果腓骨累及,亦發生同樣變化。
檢查
先天性脛骨假關節應該做哪些檢查?
本病主要是進行X線檢查:X線片可見脛骨中、下1/3前彎、成角、纖維囊性變和假關節形成,骨端變細呈錐形,骨端硬化髓腔閉鎖,骨皮質變薄,骨萎縮,脛骨遠端關節面可變形,腓骨可同時有假關節改變或彎曲畸形,小腿短縮。
治療
先天性脛骨假關節應該如何治療?
先天性脛骨假關節的治療方法頗多。Body曾統計167例手術病人,其方法有23種,方法如此之多,也正說明目前尚無一種最理想的治療方法。
1、新生兒期的治療先天性脛骨假關節根據臨床經驗,大部分在生後即可發現小腿彎曲的特有症狀,直到一次外傷或者一次錯誤的治療才形成真正的假關節。所以能在新生兒時期給以及時正確的護理和治療,無疑能提高治療效果。與此相反,一方面由於家長忽視不來就診,一方面由於錯誤診斷導致治療上的錯誤,因此臨床所見病例大部分己形成典型的假關節。生後發現單側小腿彎曲的患兒應緊密隨訪,包括定期X線檢查,觀查病情變化,即使可疑病例也應如此,直至否定該病為止。同時應向家長介紹該病的發展過程和預後及治療困難,需多次手術等基本知識,使之能與醫生緊密合作。
 石膏
石膏(2)囊腫刮除植骨術:先天性脛骨假關節囊性改變尚未形成假關節者,應及早行囊腫刮除松質骨植骨。本手術最大優點是脛骨原有的骨性支持作用存在。不需任何內固定。
2、較大兒童期的治療為假關節切除植骨術。對己經形成假關節的病例手術,目的主要是消除假關節達到骨性連線。關於手術年齡仍有不同意見;有人認為年齡越大越好,甚至提出在青春期進行手術。理由是手術成功的機會較高。但是年齡越大,畸形越嚴重,骨質吸收肢體短縮也越嚴重。有人主張5~6歲進行手術。但有人提出年齡越小手術結果越好,肢體短縮和畸形較輕,假關節癒合後提早負重會減輕生長的受限的狀態,並能促進正常骨質的發育。因此可以認為只要患兒能承受手術的打擊,就可進行該項手術。在此時期治療重點是儘快消除假關節達到堅強的骨性連線,減少再骨折。為此許多人提出了很多的手術方法,然而仍未完全解決再骨折,感染和穩定骨折斷端困難的全部問題。一般成功的病例中,有大部分都是經過兩次以上的手術。手術後均需有堅強的外固定3~6個月左右,現將幾種有代表性的手術方式簡述如下:
(1)雙重骨片植骨術:該方法由Body首倡,切除錯構組織,剪除兩端硬化的骨端,打通閉塞的髓腔,再以兩片較大的骨塊用螺絲釘固定於骨斷端兩側,其優點是避免纖維組織再度充填假關節處,以壓迫骨質影響骨質修復。該方法是比較好的方法之一,但最大的總是需要兩塊較大的骨塊為其缺點。
手術操作要點:①骨端對位後避免向前成角彎曲,並且以稍向後成角屈曲約15-20度為最好。②兩塊骨放於內外側。③勿損傷脛骨遠端骺板。
 先天性脛骨假關節
先天性脛骨假關節(3)短路植骨術:由McFarland設計提倡,畸形不必糾正,儘量用大植骨塊移植於脛骨成角的凹面,距假關節較遠的位置嵌插入骨皮質內。因植骨塊距離假關節處較遠,避免吸收而獲得癒合。經遠期觀察發現,原來畸形會自行矯正至接近正常。此法主要是由於排除了成角剪力而使假關節獲得癒合。但有時因成角畸形嚴重,負重線仍通過踝關節前面,所以在移植骨塊下端插入脛骨的地方仍會出現另一假關節。
(4)髓內外植骨術:切除骨端硬化部分,打通髓腔。以腓骨塊髓內嵌插植入,以髓內針固定,周圍以松質骨小骨塊充填。這是較經常用的一種植骨方法。
(5)帶血管腓骨移植術:顯微手術技術迅速發展而廣泛套用於外科各領域,在先天性脛骨假關節的治療中帶血管腓骨植骨己被採用,這就提高了成功率,是目前較好的治療方法之一。但再骨折形成假關節常有發生,尤其在骨遠端融合處。
(6)髓內外松質骨植骨鋼板內固定:天津醫院多採用松質骨植骨及鋼板固定術,癒合率較高,避免患兒或患兒父母切取大塊骨片所造成的後果。發現假關節處己發生骨性連線應及早手術取出鋼板和螺釘。一方面可再補充植骨,加強其堅固性:一方面可避免或減少再骨折。這種病兒術後帶鋼板負重,常常因輕度外傷造成下面螺絲釘處骨折。若及早取掉鋼板則可減少或避免再骨折的發生。
1.治療學概況本病的治療至今仍是健康搜尋一個難題健康搜尋可以採取的手術方法很多健康搜尋如大塊外置植骨複合組織瓣移植搭橋植骨、雙外置植骨等但效果均不滿意,往往植骨被吸收而發生再骨折。隨著顯微外科的發展,已開始採用吻合血管的腓骨移植或帶血管蒂的腓骨轉移鵻由於改善了局部的血液供應使本病的療效有了提高但遠期效果尚有待總結。
在治療過程中有反覆進行多次手術也達不到骨折癒合健康搜尋的效果出現下肢短縮,以致造成殘廢包括截肢的可能,這點必須充分向患兒家長說明。
對尚未形成假關節而僅有脛骨彎曲者進行手術矯正畸形是禁忌的一旦手術必將形成假關節健康搜尋,造成不堪構想的後果。
 先天性脛骨假關節
先天性脛骨假關節對於已形成假關節者手術在6~7歲之後進行比較適宜因年長兒骨骼較幼兒粗而堅硬健康搜尋手術時可取足量的骨松質及足夠長健康搜尋的骨板對骨折癒合有保證在等待手術時套用有確實作用的支具保護防止彎曲加重及骨折發生對具有較好健康搜尋的骨癒合條件者也可儘早手術。
2.幾種常用的手術
(1)Boyd手術:此手術採用健側脛骨骨板,剝去其上骨膜留有部分骨松質用健側的脛骨骨板固定於假關節處健康搜尋,再從髂骨取下骨松質植入固定。
手術是在假關節處做一縱形切口,顯露出假關節;切除所有增厚的骨膜和周圍纖維組織鵻直至健康的肌肉和皮下組織;切除假關節病變骨質,以出現骨髓腔為宜;必要時要鑽通髓腔。此步驟鵻是手術成功的關鍵健康搜尋。
此後,在假關節脛骨內側準備好植骨創面修整成平面,最大限度地使植入骨板與脛骨緊密相接植骨儘量多放於遠側端但不可損傷骨骺板整個骨板越長越好,為保持長度健康搜尋,脛骨上、下端可有少許空隙腓骨根據脛骨病變切除長度可切除一部分或火罐網不切除,保持完整腓骨能增加脛骨的穩定性火罐網。然後,上下端各用2枚螺釘固定脛骨植入骨板健康搜尋,火罐網在移植健康搜尋的骨板中間或脛骨上下端空隙中植入多量的骨松質(圖1)。縫合皮下組織及皮膚鵻為保持局部血運不要縫合深筋膜。術後用長腿石膏固定火罐網對於肥胖患兒可用單髖人字石膏固定鵻10~14天拆線,再更換石膏固定4~6個月。拆除石膏後仍套用長腿支架火罐網直至骨骼成熟雖有骨性連線但易產生再骨折,因此外固定甚為重要
 先天性脛骨假關節
先天性脛骨假關節(2)Sofield手術:此手術適用於脛骨假關節遠側端脛骨過短者。於脛骨前方縱形切開鵻顯露脛骨假關節上下端充分切除脛骨上下端病變火罐網的軟組織及骨質,應注意健康搜尋不應損傷脛骨下端的骨骺鵻擴大骨髓腔於脛骨上端截斷脛骨健康搜尋將截下的脛骨顛倒使其上端對準脛骨遠側端火罐網,用髓內針固定。若脛骨中間有空隙鵻,可取對側腓骨進行植骨使上下端緊密接觸並以有一定健康搜尋的壓力為佳(圖2)http://www.huoguan.com。縫合骨膜皮下組織及皮膚。術後套用長腿石膏固定3~6個月火罐網,但此中間可以負重刺激骨生長
 先天性脛骨假關節
先天性脛骨假關節(3)游離腓骨移植術健康搜尋:近年來健康搜尋由於顯微外科的發展健康搜尋套用健側帶血管蒂的腓骨移植取得了一定健康搜尋的效果此手術要求健康搜尋在手術顯微鏡下進行鵻,由專業的顯微外科醫生參加方可完成手術年齡鵻在6~7歲時成功率較高小齡患者鵻的手術成功率較低。術後保持吻合血管的暢通是手術成功的關鍵
(4)Ilizarovhttp://www.huoguan.com一次加壓健康搜尋一次延長術:徹底切除假關節病變部分,包括硬化骨假關節之間的纖維組織病變骨膜儘量使骨髓腔顯露出來火罐網從患兒髂骨取下骨松質塊剪成骨柴及骨條,將骨條插入髓內周圍植入少許骨柴健康搜尋套用Ilizarov外固定支架端端加壓固定鵻假關節上方再置以Ilizarov外固定支架火罐網行乾骺端骨皮質截骨,對短縮肢體進行延長(圖3)。於術後第7天開始每天延長0.5~1mm,可分2~4次進行每次延長0.25mm,不能過急,否則會造成骨不癒合一般可延長4~12mm待延長長度達到要求後即停止延長假關節完全癒合後即可去除外固定支具
 先天性脛骨假關節
先天性脛骨假關節鑑別
 先天性脛骨假關節
先天性脛骨假關節1.骨折不癒合 小兒外傷性脛骨骨折,畸形癒合可以發生,而骨折不癒合極為罕見。即使產生不癒合,骨折局部會有大量骨痂形成。
2.脆骨病 該病是全身性疾患,有多次骨折歷史。雖易骨折然骨折修復並無障礙,除此以外,該病還有特殊症狀如鞏膜發藍、聽力障礙、第二性徵早期出現及家族遺傳史。
3.佝僂病 四肢長管狀骨均有變化,下肢因負重引起膝內翻畸形多為雙側性。X線表現乾骺端變寬,骺線增寬,且有杯狀典型改變。佝僂病治癒可遺留脛骨內翻畸形。X線表現骨幹變粗,脛骨內側骨皮質有增厚。但無明顯骨質硬化,髓腔通暢。
預防
先天性脛骨假關節應該如何預防?
本病為先天性疾病,無有效預防措施,早診斷早治療是本病的防治關鍵。
但本病需注意,治療方法因人而異,不能千篇一律;對嬰幼兒以採用保守療法為好,不要輕易行截骨矯形,取病理或早期刮除植骨,過激的手術治療將導致嚴重後果。
併發症
先天性脛骨假關節可以並發哪些疾病?
一、疾病本身引起的併發症:
一旦形成假關節,患兒即不能負重行走。時間越久症狀越嚴重。小腿畸形加重有時呈現"連枷狀",甚至有些病例因骨折端刺破皮膚發生感染。
二、術後併發症:
(1)由於本病的患兒治療時需全麻,全身麻醉後常發生噁心,嘔吐,麻醉時間越長,嘔吐的發生率就越高。因此全麻病兒未清醒前去枕平臥位,頭偏向一側,防止因嘔吐引起誤吸。
(2)小兒對失血耐受力差,失血過多時常表現為面色蒼白、傷口滲血多,此時應給予及時的對症治療。

