
疾病概述
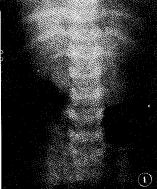 軟骨發育不全影像
軟骨發育不全影像“軟骨發育不全”這一術語來自希臘,意思是“沒有軟骨形成”,儘管患病個體有軟骨。在正常胎兒發育時期和孩童時期,除鼻和耳朵這些少數部位外,軟骨會正常發育成骨。而軟骨發不全患者卻在這個發育過程中出現一些異常,特別是在長骨(比如上臂和大腿),長骨生長板處的軟骨細胞轉變成骨細胞的速度很慢,導致了長骨短縮的發生。
軟骨發育不全患兒軀幹長度相對正常,而手臂和腿較短。上臂和大腿比前臂和小腿更短。通常,頭顱較大、前額突出和塌鼻樑也是其特徵。有時,大頭顱是腦積水的表現,往往需要手術治療。還有手短,手指粗短,中指和無名指之間分離(三叉手)等特徵。軟骨發育不全是一種常染色體顯性遺傳病,通過軟骨內分化缺陷影響長骨的發育,病人表現出特徵性的短肢性體儒,外科手術是目前有效的治療方法,基因治療是未來的發展方向。
疾病原因
1、病因
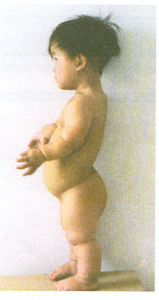 軟骨發育不全
軟骨發育不全由於發生缺陷使軟骨內骨化失敗為本病原因.在胎兒第二個月末明顯可見。90%的父母是正常的,但母親年齡大與軟骨發育不全的發病率增加有相關因素.男性軟骨發育不全型侏儒根據門德爾顯性遺傳定律遺傳已被熟知,追蹤其家族遺傳有達6代者。
軟骨發育不全為常染色體顯性遺傳性疾病有很大一部分病例為死胎或在新生兒期即死亡,多數患者的父母為正常發育,提示可能是自發性基因突變的結果。分子遺傳學研究發現系編碼成纖維細胞生長因子受體的基因發生了點突變,位置在第4對染色體的短臂上。
軟骨發育不全是由定位在4號染色體上的一個異常基因所引起的(人類有23對染色體)。有些病例,是小孩從一個患有軟骨發育不全症的父母親遺傳下來的。如果父母親一方患病,另一方未患病,那么他們的小孩患病的幾率是50%;如果父母雙方均患病,那么他們的小孩有50%的機會患這種病,25%的幾率不患病,還有25%的幾率從每一父母親那遺傳一個異常基因,並且有嚴重的、導致早死的骨骼異常。未遺傳異常基因的小孩將完全不會有該病的影響,並且也不會把它傳遞給自己的子女。然而,超過80%的病例可以不是通過遺傳得來的,而是由形成胚胎的卵細胞或精細胞內的新突變所引起的。由這種新突變所引起的患兒,其父母身材可以是正常的。這種情況的典型現象是:這些父母只有這一個患病小孩,並且他們第二次再懷孕同樣疾病的小孩的風險特別小。遺傳學家已經發現高齡父親(40歲和40歲以上)生育軟骨發育不全症或由新突變引起的某種其它常染色體顯性遺傳病患兒的幾率更高。如前面所述,由新突變引起的軟骨發育不全症患病個體將把這種疾病傳遞給他們的後代。
2、病理改變
 軟骨發育不全--CT
軟骨發育不全--CT軟骨發育不全為骨骺盤的軟骨增生帶出現異常,缺乏軟骨形成.看不到正常軟骨細胞的柱狀排列。並發生粘液樣退行性改變.海綿骨中的骨小梁不規則,但鈣化正常,骨長度生長速率很低。由於膜內化骨的作用正常,故骨的直徑發育正常,但因長度不足而顯得增粗.偶爾見到血管中的結締組織增生形成橫行纖維帶使骨骺早期癒合而影響長度發育停止。
臨床表現
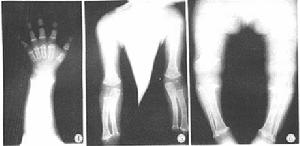 軟骨發育不全
軟骨發育不全(二)頭顱增大有的病人有輕度腦積水,穹隆及前額突出,馬鞍型鼻樑,扁平鼻、厚嘴唇舌伸出(在嬰兒)。
(三)胸椎後突,腰椎前突,以後者為明顯。骶骨較水平使臀部特徵性的突出。
(四)胸腔扁而小,肋骨異常的短。
(五)手指粗而短,分開,常可見4、5指為一組,2、3指為一組,拇指為一組,似“三叉戟”。有的病人的伸肘動作輕度受限。
(六)下肢呈弓形,走路有滾動步態(rolling)。
(七)智力發展正常,牙齒好,肌力亦強,性功能正常。
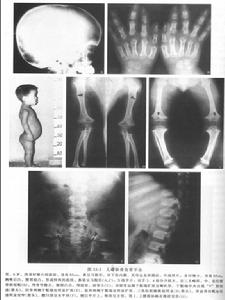
X線表現①顱蓋大,前額突出,頂骨及枕骨亦較隆突,但顱底短小,枕大孔變小而呈漏斗型,其直徑可能只有正常人的1/2。如伴發腦積水側腦室擴張。②長骨變短,骨幹厚,髓腔變小,骨骺可呈碎裂或不齊整。在膝關節部位,常見骨端呈“V”形分開,而骨骺的骨化中心正好嵌入這V形切跡之中。由於骨化中心靠近骨幹,使關節間隙有增寬的感覺。下肢弓形,腓骨長於脛骨,上肢尺骨長於橈骨。③椎體厚度減少,但脊柱全長的減少要比四肢長度的減少相對少很多。自第一腰椎至第五腰椎,椎弓間距離逐漸變小。脊髓造影可見椎管狹小,有多處椎間盤後突。④骨盆狹窄,骼骨扁而圓,各個徑均小,髖臼向後移,接近坐骨切跡,有髖內翻,髖臼與股骨頭大小不對稱。肋骨短,胸骨寬而厚。肩胛角不銳利,肩胛盂淺而小。
疾病檢查
 軟骨發育不全示意圖
軟骨發育不全示意圖由於肋骨短的緣故,胸腔的前後徑變小。胸骨短,寬並增厚,脊柱高度正常,但其骨化核較小,常有輕度駝背,腰椎的椎弓根間距離小於正常,椎弓根短而粗,椎體與椎弓可早期癒合而形成椎管狹窄,由於蝶骨前後及枕骨基底的生長受阻。枕骨大孔小於正常並呈漏斗狀。顱骨其餘部分則發育正常,故顯得前額突出。
本病主要是進行X線檢查,其表現主要有以下幾點:
①顱蓋大,前額突出,頂骨及枕骨亦較隆突,但顱底短小,枕大孔變小而呈漏斗型,其直徑可能只有正常人的1/2。如伴發腦積水側腦室擴張。
②長骨變短,骨幹厚,髓腔變小,骨骺可呈碎裂或不齊整。在膝關節部位,常見骨端呈“V”形分開,而骨骺的骨化中心正好嵌入這V形切跡之中。由於骨化中心靠近骨幹,使關節間隙有增寬的感覺。下肢弓形,腓骨長於脛骨,上肢尺骨長於橈骨。
③椎體厚度減少,但脊柱全長的減少要比四肢長度的減少相對少很多。自第一腰椎至第五腰椎,椎弓間距離逐漸變小。脊髓造影可見椎管狹小,有多處椎間盤後突。
④骨盆狹窄,骼骨扁而圓,各個徑均小,髖臼向後移,接近坐骨切跡,有髖內翻,髖臼與股骨頭大小不對稱。肋骨短,胸骨寬而厚。肩胛角不銳利,肩胛盂淺而小。
疾病治療
一般不難,在水典型的病例,需與其他原因所引起的侏儒區別。①軟骨發育欠全(hypochondroplasia)侏儒表現不太明顯,頭顱正常。②軟骨-外胚層發育不全(chondro-ectodermaldysplasia),即EllisVan-Creveld綜合徵,為短肢型侏儒,伴有胸部畸形和心臟病變,並指、指甲牙齒髮育不良。肢體縮短的部位常發生在遠段骨骼。③脊柱-骨骺發育不全(spondylo-epiphysealdysplasia)。亦為短肢型侏儒,常有近端大關節的破壞,顱骨正常,脊椎椎體變扁,椎體骨化中心互相吻合。胸廓發育不良如鈴形。④佝僂病及克汀病。佝僂病有典型的臨床及X線表現,容易區別;而克汀病常伴有智力發育不良。方1青果酒
組成:青果50克,白酒500克。
用法:青果洗淨,置瓶中,加入白酒,密封3周,分次飲用,每次10~15克。
功效:行氣活血止痛。
主治:軟骨發育不全,持續復發局部壓痛,捫及增生組織者。
附註:青果有行氣散寒止痛之效。
來源:民間驗方。
方2茯苓紅棗粥
組成:茯苓粉30克,紅棗15枚,粳米150克。
用法:紅棗洗淨,加水煮至爛;粳米煮粥,待粥將成時倒人紅棗及湯,加入茯苓粉,再文火煮20分鐘,加少許紅糖,趁熱服用。
功效:活血消腫。
主治:軟骨發育不全,局部腫脹疼痛顯著者。
來源:夏翔,等.家庭食養食補食療全書.瀋陽:遼寧科學技術出版社,2001,736
方3鮮香椿拌豆腐
組成:鮮香椿100克,豆腐1塊,精鹽5克,香油5毫升。用法:鮮香椿洗淨開水燙一下,冷卻後切成碎末,放在豆腐上,加入香油、鹽後拌勻食用。
功效:緩急止痛。
主治;軟骨發育不全,疼痛急性發作,局部觸痛,組織增厚者。
附註:鮮香椿味苦性平,消炎止痛。
來源:夏翔,等.家庭食養食補食療全書。瀋陽,遼寧科學技術出版社,2001,736
方4海帶茴香湯
組成:海帶15克,海藻15克,小茴香6克。
用法:海帶、海藻、小茴香分別洗淨,置鍋中,加清水500毫升,煮開5分鐘,去茴香,喝湯,連續服用10天。
功效:清熱止痛,軟堅。
主治:軟骨發育不全,肋軟骨處壓痛、增生者。
附註:海帶軟堅化痰,利水泄熱;海藻清熱解毒,軟堅散結。
來源:夏翔,等.家庭食養食補食療全書.瀋陽:遼寧科學技術出版社,2001,159
方5金橘米酒
組成:金橘1000克,米酒500毫升。
用法;金橘洗淨,置瓶中,加米酒,密封l周后,分次飲服。
功效:健脾理氣止痛。
主治:軟骨發育不全伴脾胃不和,胸肋壓痛,胃納差者。
來源:夏翔,等.家庭食養食補食療全書。瀋陽:遼寧科學技術出版社,2001,97
方6佛手香薷飲
 橘皮米粥
橘皮米粥用法:將佛手、香薷分別洗淨,切成片,置鍋中,加清水500毫升,急火煮開3分鐘,加白糖,分次飲服。
功效:行氣止痛。
主治:肋軟骨炎疼痛不愈數月,復發,軟骨發育不全,局部增生者。
附註:佛手行氣止痛,香薷化濕利水。
來源:民間驗方。
方7茄子根酒
組成:茄子根100克,白酒500毫升。
用法:茄子根洗淨,置瓶中,加白酒,密封3周,分次飲服。
功效:清熱消腫止痛。
主治:肋軟骨炎疼痛劇烈,軟骨發育不全,局部刺痛,咳嗽尤劇者。
附註:茄子根有清熱消腫止痛作用。
來源:民間驗方。
方8橘皮米粥
組成:橘皮30克,粳米50克。
用法:將橘皮洗淨,曬乾,碾為細末,粳米加清水500毫升,置鍋中,急火煮開5分鐘,加橘皮細末,文火煮30分鐘,成粥,趁熱食用。
功效:行氣止痛,健脾開胃。
主治:軟骨發育不全伴脾胃不和者。

