症狀體徵
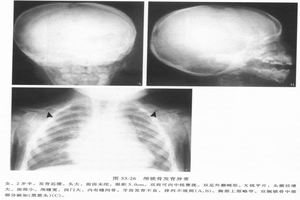 多骨纖維發育不良
多骨纖維發育不良 多骨纖維發育不良
多骨纖維發育不良3.性早熟以女性多見,性早熟的年齡多在6歲以前,平均發育年齡為3歲。有出生後第1個月就出現性早熟的報導。女性月經來潮是性早熟的首要症狀,發生在乳腺發育前,血漿雌激素水平波動在正常或顯著升高(>900pg/ml)之間,常呈周期性。幼年女性的LH和FSH水平受到抑制,並對GnRH刺激無反應(除成人患者),說明女孩的性早熟並不依賴促性腺激素。女性性早熟與骨齡相一致。長效的GnRH類似物賽庚啶(cyproheptadine)對青春期前病人治療無效.而芳香酶抑制劑睪內酯對女孩有明顯的治療作用。男孩出現的性早熟比女孩要少得多。睪丸增大是相對稱的,隨後出現陰jing增大和陰毛,與正常青春期相等。睪丸的組織學檢查證實曲細精管增大,沒有或有少量的間質細胞。在女孩和男孩中,當骨齡達到青春期年齡時,促性腺激素分泌開始,對GnRt的反應變為青春期,此時為促性腺激素依賴性性早熟便與早先的非促性腺激素依賴性性早熟重疊,在女孩中月經多變得規律,在成人階段,女性病人的青春發育期正常,有正常的生殖功能。
 病因
病因(1)甲狀腺異常:可發生在MAS的任何年齡,甚至在出生後不久就發生。發生本症的甲亢與Graves病的特徵不同,在男女中的分布相同,其出現甲狀腺腫趨於多發性結節。結節常為良性,顯示放射性碘攝取率增高。多數情況下,結節常並發甲亢,但測定甲狀腺激素常在正常上限。TRH刺激試驗顯示,TSH釋放受抑,甲狀腺病理顯示無淋巴細胞浸潤,也無甲狀腺抗體。這些均可與臨床的嚴重甲亢相鑑別,如果甲亢臨床明顯,也可用抗甲狀腺藥物、同位素或手術治療。
(2)皮質醇增多症:在MAS中並不如甲亢和性早熟常見。兒童皮質醇增多症表現為生長率減低,MAS發生的皮質醇增多症出現的年齡可能很早且症狀嚴重。ACTH水平較低,大劑量地塞米松不能抑制其腎上腺功能。
(3)高磷酸尿和低磷血症佝僂病或骨質軟化可與MAS合併存在:MAS患者尿cAMP增加,與腎小球cAMP的濾過增加有關。若尿磷反應正常,尿cAMP對輸入外源性PTH的反應遲鈍。用維生素D治療和口服磷酸鹽,佝僂病和軟骨病通常能得以糾正,但多數對治療有抵抗。
(4)其他:肝臟的異常包括嚴重的新生兒黃疸,肝臟的酶活性增加,肝活檢時發現膽汁淤積和膽管異常的表現。心臟的異常包括心臟擴大,持續心動過速和在年輕患者中的突發死亡。非典型心肌細胞肥厚在組織學上也表明內分泌異常的作用。限制性肺疾患、動靜脈分流或心臟原發異常和心臟傳導異常的病因還不清楚。其他與MAS有關的異常未見報導。
疾病病因
病因未完全闡明。但近年來有突破性進展。本徵患者廣泛存在著鳥核苷酸結合蛋白(G蛋白)的興奮性α亞基(Gs)基因突變,後者導致前成骨細胞增殖和骨纖維發育不良。Gs突變多位於基因編碼的RaolC或RaolH部位。病理生理
 發病過程
發病過程新近又有學者通過逆轉錄聚合酶鏈反應技術用特異的等位基因引物、特異等位核苷酸雜交和DNA排序,測得Gs的α亞單位的突變位於基因編碼的R201C或R201H部位。而引起突變的原因是因為GTP酶的活性增高,損傷了介導的受體活性所致。
診斷檢查
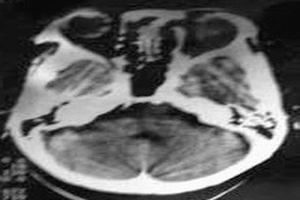
 檢查
檢查具有下列第1項或第2項則診斷可成立,如具有第3項應高度考慮MAS診斷的可能。
1.具有骨損害、皮膚色素沉著和性早熟3大主征。
2.具有骨纖維結構不良的X線表現,皮膚Cafe-au-lait點,年齡在30歲以下,伴有內分泌或非內分泌的異常。
3.具有第1項中的兩種或一種,加上內分泌或非內分泌異常的年輕患者。
實驗室檢查:
生化或放射免疫檢查,其改變與合併內分泌異常有關,如合併甲旁亢,則血鈣可升高,尿磷增多,血磷降低,ALP增高。合併性早熟,血清雌、孕或雄激素水平增高。合併肢端肥大症和高泌乳素血症則可測得增高的GH或PRL等。因病程緩慢,個別病情輕者也可無生化異常。
其他輔助檢查:
1.X線檢查MAS的骨組織被異常增生的纖維組織所取代,表現為不同程度的骨膨脹,骨皮質變薄,但完整。骨密度可為均勻性減低,或呈磨玻璃樣,或見條索狀、斑點緻密陰影。國內總結X線分4型:
(1)囊狀改變:分單囊和多囊,邊緣硬化,囊內常見條狀骨及斑點狀緻密影,常見於管狀骨和肋骨。
(2)毛玻璃改變:髓腔囊狀膨脹呈毛玻璃狀密度,內可有條狀骨紋和斑點狀鈣化。
(3)絲瓜筋狀改變:骨小梁粗大扭曲,頗似絲瓜筋狀,嚴重者如蛛網狀,長管狀骨粗大,骨狀紋一般和縱軸平行。
(4)蟲蝕狀改變:單發或多發的溶骨性改變,邊緣銳利如蟲蝕樣,可類似溶骨性轉移性破壞。
此外,脊柱和長骨常伴病理性骨折。
2.CT及MRI檢查CT檢查費用雖較X線照片高,但其密度分辨高於X線、對病骨內的囊變、破壞、鈣化和骨化顯示較X平片敏感準確。CT橫斷面克服了常規X平片前後重疊的缺點,可用於頭顱、脊柱和骨盆等重疊較多的部位。MRI對MAS的病理顯示無疑較常規X線或CT更敏感,能顯示大部分在X線平片或CT片上不能顯示的病灶(如壞死、液化、出血),纖維或纖維骨樣組織病灶在T1加權像和T2加權像均呈低信號。骨幹結構不良在病變的不同階段可有不同的病理改變。如病灶內的壞死液化在T1加權像上呈低信號,在T2加權像上呈高信號。如壞死組織合併出血,T1加權像上呈高信號。病灶內的鈣化和周緣的硬化在T1加權像和T2加權像上呈明顯的低信號。此外,少數病灶邊緣在T1和T2加權像上呈薄帶狀環狀高信號,其病理機制不清。某些病灶在T1加權像上呈不均勻的中低信號,而在T2加權像上則平片所見的“絲瓜筋”樣纖維結構不良。而液化的病灶在T1加僅像上為中低信號,在T2加權像上為均勻高信號。
鑑別診斷
 辨別相似疾病
辨別相似疾病主要與變形性骨炎和神經纖維瘤病(vonRecklinghausen病)鑑別。
1.變形性骨炎MAS骨病不典型時易與變形性骨炎相混淆,但變形性骨炎無性早熟,亦無色素沉著的Cafe-au-lait斑點,而血ALP明顯升高,可資鑑別。
2.神經纖維瘤病累及骨骼,常合併有皮膚咖啡斑,不合併內分泌異常,可與MAS類似。
神經纖維瘤病有皮下結節或軟性色塊改變及多發性神經纖維瘤,亦無性早熟。
治療方案
 藥物治療
藥物治療1.骨損害的內科治療
(1)降鈣素:降鈣素50~100U或益鈣寧40U隔天或1周2次,肌注,有人認為該藥對骨畸形造成的局限腫脹和骨折刺激神經末梢引起的疼痛有明顯止痛作用,無明顯副作用,因價格昂貴影響長期用藥。
(2)二磷酸鹽製劑:療效各家報導不一,常用的有:
①EHDP(disodiumedetate):每天20mg/kg,口服療程6個月~1年。
②帕米磷酸鹽(pamidronate):60mg/d,靜脈滴注連用3天,每6個月重複1次。病人在治療期間無自發性骨折發生,在治療2~3療程後骨痛及步態異常消失,肢體長度無變化,血ALP、尿羥輔氨酸下降,但尚未得到骨受損治癒的放射學及閃爍照相的證實。以上兩種藥使用也未見明顯副作用(可能與病例觀察太少有關),作用機制尚未明了。
2.骨畸形的外科治療對於MAS的肢體畸形嚴重者可行截骨矯形術。刮除病灶骨,採用植骨與內固定,術後有可能復發。若病變迅速增長,要警惕惡變為骨纖維肉瘤,其次是骨肉瘤,此時不宜放射治療,以免誘發惡變。
3.性早熟的治療
(1)羥孕酮(MPV):5~10mg/d,可抑制FSH、LH分泌,使乳房回縮,月經停止,或使陰jing和睪丸縮小,陰毛減少,療效肯定。少數病人有噁心、嘔吐、乏力、嗜睡等副作用,有肝、腎功能不全者慎用。
(2)醋酸甲地孕酮(danazol,danacrin,達那唑):能抑制促性腺激素的分泌高潮,不抑制正常體內FSH、LH的基礎水平,療效同上,常用量50mg/次,1~2次/d或酌情調整劑量。副作用為肝臟轉氨酶升高,長期套用需定期檢查肝功能,肝功能不全者禁用大劑量。
(3)甲羥孕酮(depoprovera):每次100~200mg,肌注,每2~3周1次,作用、療效與MPA類似。
(4)酮康唑:具有抑制腎上腺和性腺類固醇合成的作用。最近來自美國馬里蘭醫療中心的Syed和chalew的報導顯示,他們使用酮康唑200mg,3次/d治療2例MAS的女性性早熟的病人1年後,性早熟得到遏止。副作用是皮膚瘙癢、皮疹,但並不影響使用,肝功能異常停藥後可恢復。
(5)睪酮內酯:能抑制雌激素分泌,促進女孩乳腺成熟。也促進骨的線性生長和骨骼的成熟。對非促性腺激素依賴性性早熟療效肯定。用量為20~40mg/(kg?d)。
(6)他莫昔芬(tamoxifen):是目前認為最優秀的抗非促性腺激素依賴性性早熟的藥物,該藥能與雌二醇競爭雌激素受體,使雌激素耗竭。用此治療後,青春發育減慢,線性生長下降,當中樞性青春期發育時,給予GnRH類似物治療,生長速度及骨骼成熟維持穩定。用量為10~20mg,2次/d。
併發症
 併發症
併發症
