
簡介
 桿狀體肌病
桿狀體肌病類型
該病按臨床特徵可分為4個類型。
①先天性急性致死型:該型出生即出現全身肌無力及肌張力低下,以肩胛帶及骨盆帶肌受累明顯,常伴脊柱側彎等畸形,多因呼吸衰竭而死亡;
②先天性相對穩定或緩慢進展型;
③亞臨床型:該型無明顯臨床表現,僅在肌肉活檢時發現異常;
④成年起病型。
病因
 桿狀體肌病
桿狀體肌病①TPM3基因定位於染色體1q21~23,編碼α-原肌球蛋白(α-tropomyosin),呈常染色體顯性或隱性遺傳,見於新生兒發病患者;
②NEB基因,位於2q21.2~22,編碼nebulin,呈常染色體隱性遺傳;
③位於1q42.1的ACTA1基因,編碼骨骼肌α-肌動蛋白(α-actin),呈常染色體顯性或隱性遺傳,10%~20%桿狀體肌病是由ACTA1基因錯義突變引起的,大多數患者是散發病例,但其父母可查見原發突變;
④TPM2基因,位於9q13.2,編碼β-原肌球蛋白(β-tropomyosin),屬常染色體顯性遺傳;⑤TNNT1基因位於19q13.4,編碼Ⅰ型肌纖維肌小節細絲的肌鈣蛋白T1。
病理
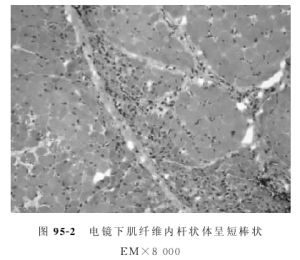
肌肉活檢HE與MGT染色均能發現位於肌膜下、肌原纖維之間或核內的大量短棒狀結構,又稱為桿狀體,MGT染色呈紫紅色。桿狀體本身無酶活性,ATP酶染色不著色。
肌肉病理改變除桿狀體結構外無其他異常發現,心肌中也可找到桿狀體結構。Ⅰ型或Ⅰ、Ⅱ兩型纖維均可出現桿狀體,常伴隨Ⅰ型纖維占優勢,Ⅱ型纖維減少、ⅡB型纖維缺乏現象。不同肌肉組織的桿狀體數量不一致,與病情嚴重程度無關。患病家族中無症狀者可僅出現選擇性Ⅰ型纖維占優勢,而無桿狀體結構。
電鏡下桿狀體呈短棒狀緻密結構,與Z盤分布一致。病情嚴重患者可發現核內桿狀體。

