
概述
胼胝體發育不全(dysgenesisofthecorpuscallosum,DCC)屬先天性顱腦畸形,包括胼胝體部分缺如或全部胼胝體和周圍結構的缺如。臨床無特殊症狀,重者可有智力障礙、癲癇和顱內壓增高症狀,甚至呈痙攣狀態。
病因
胼胝體是大腦兩半球問最主要的一大塊有髓纖維的集合體,連線著兩側大腦半球,並形成側腦室的頂。它是從原始終板發生的前腦聯合之一。
胼胝體形成於胚胎的第12~20周。胚胎第7~10周時,終板背側普遍性增厚,其上方形成聯合塊,後者誘導大腦半球軸突從一側向另一側生長,形成胼胝體。
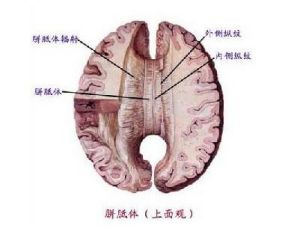 解剖學示意圖
解剖學示意圖如果聯合塊不能誘導軸突從大腦半球一側越過中線到達對側大腦半球,則胼胝體就不能形成。胼胝體分為嘴部、膝部、體部和壓部4個部分,其發育順序由前向後,正好與其成熟順序相反。胚胎早期的宮內感染、缺血等原因可使大腦前部發育失常,而發生胼胝體缺失,晚期病變可使胼胝體壓部發育不良。常首先累及體部和膝部,也可同時累及膝部和壓部,但單獨累及膝部的較少,僅見於前腦無裂畸形。但Barkovich(1988)認為胼胝體發育不良,是由於胼胝體形成的前驅階段受損,並非發生於胼胝體形成期。胼胝體發育不良也有遺傳基礎。
病理
胼胝體發育不良或缺如自18l2年Rell進行了屍解報告以來,此後Bull(1967)Brun(1973)等也對其進行了詳細描述。胼胝體發育不良可為完全或部分缺如。景常見的是胼胝體和海馬連合完全性發育不良,而前連合得以保留。在胼胝體所保留的纖維束中,只有Probst束,這是向前後方向投射,不越過中線的纖維柬。由於沒有胼胝體纖維的約束力。第三腦室頂向背側抬高.室間孔明顯擴大,使第三腦室和側腦室形成一個"蝙蝠"形囊腔。側腦室后角向中間方向擴大。在胼胝體部分發育不全中.最常見的是壓部缺失,但體部和嘴部的任何一部分均可受累。
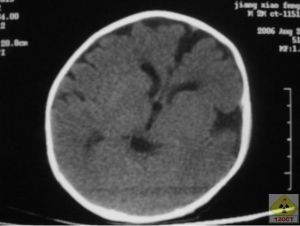 CT圖
CT圖臨床表現
胼胝體發育不良大多數為散發性,原因不明。但也有在姐妹中發病和兄弟中發病者,家族發病者呈X-4連鎖隱性發病。
其臨床症狀、體徵與合併的其他腦畸形有關,因為先天性胼胝體發育不全或缺如的本身一般不產生症狀。在成人患者中,用複雜的心理測定檢查方法,可發現兩半球問的信息傳遞有輕微障礙。新生兒或嬰幼兒患者可表現為球形頭、眼距過寬和巨腦畸形,多在懷疑腦積水行CT掃描檢查時,才發現有胼胝體發育不良或缺如的特徵性圖象。可出現智力輕度低下或輕度視覺障礙或交叉觸覺定位障礙。嚴重者可出現精神發育遲緩和癲癇。因腦積水可發生顱內壓增高。嬰兒常呈痙攣狀態及錐體束征。X-4性聯遺傳者,其特點為生後數小時有癲癇發作,並出現嚴重的發育遲緩。
 患病兒童
患病兒童1、縱裂接近三腦室前部(胚胎期縱裂與透明隔間腔相通,以後被胼胝體嘴封閉,若嘴不發育則縱裂與透明隔間腔相通,直達三腦室前部。嘴部發育最晚,無論胼胝體發育不全或不發育均累及胼胝體嘴部。所以縱裂與三室前部相通是最常見的表現。
2、胼胝體全部或部分缺如,部分缺如往往發生於胼胝體壓部。海馬,前或後連合缺如。
3、側腦室前角向外移位,側腦室內側緣有凹陷的壓跡。原因是原先連線兩側半球的前部胼胝體缺如,那些本來橫向連線兩側半球的纖維現在呈縱向排列,位於側腦室內緣,壓迫側腦室,形成壓跡。畸形的兩側腦室前角彼此分離,形成蝙蝠翼狀。
4、側腦室體分離,相互平行,主要見於橫斷面圖象上,可能是輕度胼胝體發育不全僅有的表現。
5、胼胝體壓部缺如,使側室三角區擴大。
6、大腦半球內側面的腦溝呈放射狀排列(在矢狀面圖像上)。
7、海馬發育低下,導致側室顳角擴大。深部白質發育不良也是側室擴大的原因。
8、第三腦室位置升高,並呈囊狀擴張,使兩側大腦內靜脈分離。
9、在兩側半球之間的縱裂中形成大的囊腫,囊腫和第三腦室是分離的,與側腦室之間可有或無交通。囊腫可以只位於大腦鐮的一側,或跨大腦鐮,位於大腦鐮兩側。
10、胼胝體膝部可合併脂肪瘤,脂肪瘤也可以延伸至胼胝體的所有部分。
鑑別診斷
胼胝體脂肪瘤CT表現典型,診斷不難,根據其CT值脂肪密度易與縱裂囊腫和皮樣囊腫鑑別,後者密度較脂肪瘤高。
胼胝體發育不良大多數為散發性,原因不明。其臨床症狀、體症與其合併的其他腦畸形有關,因為先天性胼胝體發育不全或缺如的本身一般不產生症狀。在成人患者中,用複雜的心理測定檢查方法,可發現兩半球問的資訊傳遞有輕微障礙。胼胝體發育不良可以單獨存在,甚至不產生任何症狀,但這種情形非常罕見。
絕大多數罹患此症的孩子都多多少少會有些外觀的異常,如頭太大或太小、下巴小、眼睛有斜視、白內障、或眼球過小、唇顎裂、腎臟與輸尿管異常、多指症或並指症等,多在懷疑孩子異常之情況可能為腦積水的原因所引起,進而執行電腦斷層掃描檢查,才發現有胼胝體發育不良或缺如的特徵性圖象。
胼胝體發育不良可出現智力輕度低下或輕度視覺障礙或交叉觸覺定位障礙。嚴重者可出現精神發育遲緩和癲癇。因腦積水可發生顱內壓增高。嬰兒常呈痙攣狀態及錐體束征。X-4性聯遺傳者,其特點為生後數小時有癲癇發作,並出現嚴重的發育遲緩。
胼胝體發育不良雖然在胎兒期便已形成,但出生之後,並沒有一些特徵告訴我們嬰兒罹患此症,往往是在嬰兒發展的過程中,因為動作或語言發展遲緩、癲癇症(尤其是嬰兒點頭痙攣)或腦性麻痹等神經症狀出現,做了腦部超音波或腦部磁振掃描才發現這種情況。如果說嬰兒的外型上有任何值得我們懷疑有胼胝體發育不良的可能性的話,則兩眼分離過遠與斜視這兩種情況就要讓我們警覺到此症存在的可能性。絕大多數的個案的神經發展方面也不正常,常會合併動作與語言發展遲緩、智慧型障礙、癲癇症與腦性麻痹。
藉由核磁共振或是電腦斷層等方式來做判斷是否有出現胼胝體發育不全。
治療
就目前的治療而言,已經存在的胼胝體發育不良是沒有任何方法可以改變它,也無任何藥物可以治療胼胝體發育不良。但是對於伴隨發生的癲癇問題,則可以採取藥物控制治療。預防方法
復健訓練
雖然胼胝體發育不良並無方式或藥物可以改變胼胝體本身的問題,但我們能做的是針對孩子所呈現出來的缺陷或障礙,予以積極的早期療育,越早施行各項療育,將來出現的障礙會越輕、越少。
 康復訓練
康復訓練
