疾病概述
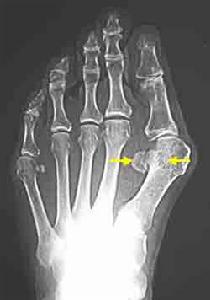 畸形性骨炎
畸形性骨炎變形性骨炎即“Paget病”。又名“畸形性骨炎”、“Paget綜合徵”。為一種原因未明的,可能為先天性骨代謝異常所致的局限性變形性疾病。為一種成人的慢性骨骼病,其特徵為骨局部代謝過強,骨組織被軟化和增大的骨性結構取代。該病是骨重建(boneremodeling)異常所致的臨床綜合徵,其病變特點是過多的破骨細胞失控制後引起高速骨溶解,並導致成骨細胞增多和骨形成過多生成的骨組織結構脆弱。骨鹽及膠原的轉換率顯著增高致使骨局限膨大、疏鬆易發生病理性骨折;骨周圍血管增生或出現骨肉瘤。變形性骨炎的病變侵蝕廣泛,全身骨骼均可受累,好發部位是股骨脛骨顱骨脊椎的腰骶部及骨盆。
本病為英國醫師Paget於1876年首次報告。病理特點為骨的破骨細胞過度活躍引起的骨溶解加速,繼之再生,伴有不規則的新骨形成。臨床上40歲以上成人多見。主要表現為局限性骨骼變形及骨痛、病理性骨折以及由於骨質過度壓迫第8對腦神經而致耳聾。血鈣正常,尿鈣排出增加。X線表現為有骨骺過渡生長與破壞交錯的傾向,溶骨區可有囊形成,長骨骨小梁增粗。在病變部位有多發性小動脈瘺,病程延長,常可並發心力衰竭、腎結石、骨折或成骨肉瘤。治療最有效的方法是長期套用降鈣素。
一種病因不明,以進行性風濕樣骨關節痛、脊柱和四肢畸形、病理骨折及腦、脊髓壓迫症狀為主要特徵的慢性骨疾病。1876年由J.佩吉特首先描述,故亦稱佩吉特氏病。又稱畸形性骨炎。發病率隨地區、種族、年齡而有很大差異,在西歐及澳大利亞、紐西蘭等地區多見,非洲、東亞(包括中國)則極少見。男女性均可累及,多發生在40歲以上者,有家族史者占15%。
疾病原因
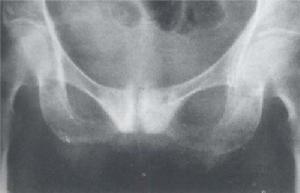 變形性骨炎
變形性骨炎曾有炎症、腫瘤、內分泌紊亂、血供異常、自身免疫及結締組織代謝先天缺陷等各種假說,但都缺乏充足證據。目前多認為本病是一種慢性病毒性感染,證據是:①超微結構觀察發現,破骨細胞的細胞核及細胞質內有典型的包涵體(可能是一種副粘液病毒的核蛋白包膜),與呼吸道合胞病毒培養細胞內的包涵體極其相似;②本病有較長的潛伏期,呈亞急性臨床過程;③本病的骨破壞及骨形成伴以纖維性變,是一種慢性炎症反應;④病變部位的大量多核巨細胞可能是多核合體巨細胞的遺留;⑤本病發病有一定地區性;⑥不少病例有家族史。
2、病理
受累骨骼富含破骨細胞和成骨細胞,骨髓為纖維結締組織所侵襲,血供豐富。總的說,其病理變化為反覆發生越來越重的骨吸收。接著出現過度修復現象,骨變成畸形,質脆,長骨可彎曲,扁骨可變形,而發生病理骨折。
3、生理
溶骨期的骨組織輕而軟,富含血管。硬化期的骨骼肥大變硬。鏡下破骨細胞和成骨細胞均顯著增多,骨髓為纖維結締組織侵襲,骨皮質和髓質分界不清,結構雜亂,呈“鑲嵌構象”(mosaicpat-tern)。病灶邊緣出現破骨性骨吸收,其後方為成骨性再生。破骨細胞顯著增大,胞核極多,有時多達100個以上,成骨細胞亦明顯增多,核大,有明顯核仁及異染色質,核緣內陷。硬化性變形性骨炎的組織病理改變可與甲旁亢類似,但破骨細胞的上述改變仍較明顯。
臨床表現
 變形性骨炎--骨結構減弱
變形性骨炎--骨結構減弱多數常無症狀,呈隱匿起病。症狀有疼痛,僵硬感,易疲勞,骨畸形,頭痛,聽力減低,頭顱增大。Paget骨痛是深部酸痛,偶為劇痛,夜間可加重。疼痛也可由神經受壓引起或與骨關節炎有關。決定於病變範圍和部位、單骨性或多骨性、有無畸形及合併症等。本病好發於中軸骨,但任何骨骼均可被累及。常有疼痛。患者常見骨畸形,如頭顱增大、駝背和四肢彎曲畸形,同時可出現顱底陷入、脊髓及神經根受壓迫症狀,還可出現耳硬化、耳聾、視神經萎縮等。
由於骨結構減弱,輕微損傷即可引起骨折,最常見於股骨、脛骨、肱骨、脊椎骨和骨盆。約1%患者可發生肉瘤。40歲以上發生肉瘤者約1/5患變形性骨炎。X射線檢查顯示,早期骨結構的變化主要為吸收破壞,以後可呈囊狀或蜂窩狀骨吸收。生物化學檢查顯示血清鈣正常或偏高,鹼性磷酸酶明顯增高,酸性磷酸酶含量也輕度增加。尿鈣及尿羥脯氨酸排泄增加。
根據以上表現可作出診斷,本病應與甲狀旁腺功能亢進、多發性骨髓瘤相鑑別。本病又可合併退行性關節炎、鈣化性關節周圍炎、痛風或心血管異常等。
2、體徵
為雙顳部顱骨增大,前額隆起,頭皮靜脈曲張,一側或雙側神經性耳聾或耳硬化症,眼底有血管樣紋,軀幹矮而駝背,形似猿猴。蹣跚步態,股或小腿前外側彎且有骨膜壓痛和溫度升高。聽覺喪失,脊髓狹窄症,不全麻痹或截癱均為神經受壓的表現。因Paget骨病是代謝活躍而且血管嚴重受累的病變,故可發生高排性心力衰竭。彎曲的長骨與鄰近關節的骨關節炎可發展為畸形。可發生病理性骨折。約1%的病人發生肉瘤變性,此時疼痛越來越劇烈。
主要有骨折腰腿痛、關節病變心、血管異常、耳聾眼和皮膚病變及高尿酸血症等。
1.骨折主要有3種類型的骨折:即裂紋骨折長骨斷裂和椎體壓縮性骨折。可在輕微外傷或無外傷情況下發生,骨折不癒合率達。
2.腰腿痛可能是繼發性小關節炎或病理性骨折所致。
3.關節病變與變形性骨炎相伴的關節病變有關節畸形、退行性關節病變、軟骨鈣鹽沉積和假性痛風、鈣化性關節周圍炎等。骨病變畸形可能導致關節畸形,但變形性骨炎本身很少侵犯關節軟骨面;當骨畸形累及髖關節相鄰部位時,因運動應力異常可導致關節異常磨損軟骨缺損,而下層出現假血管瘤樣物晚期出現髖臼內陷。膝關節也有類似情況。在遠離病灶的部位,可出現鈣化與病變的擴展無關。
4.心血管異常變形性骨炎累及骨骼達30%以上時或單獨累及顱骨時可出現心排出量增加,管鈣化阻塞導致血流量增加。重症變形性骨炎常並發心瓣膜鈣化及相關病變。主動脈狹窄達30%,完全性房室傳導阻滯、不完全性房室傳導阻滯、束支傳導阻滯和左室肥厚的發生率分別為11%、11%20%和13%,重度顱底陷入時可伴有動脈“竊血”綜合徵。
5.耳聾顱骨外板增生可引起顱底孔道變窄壓迫顱神經,其好發部位為顳骨岩部,故常合併聽神經功能障礙導致感覺性聽力喪失、中耳骨化和慢性炎症等病變,或導致視盤水腫眼肌病變、突眼,視神經萎縮及失明。
6.高鈣血症和高鈣尿症僅見於一些病變廣泛和長期不活動者此外,有些病人因尿酸過多可導致高尿酸血症,部分病人可出現腎石病、高鈣血症和高鈣尿症。可能與合併原發性甲旁亢,或與廣泛性骨損害有關。7.惡性病變變形性骨炎並發肉瘤約占1%,多數為骨肉瘤亦可為纖維肉瘤或其他類型的肉瘤,繼發性骨巨型細胞瘤少見變形性骨炎合併骨肉瘤(Paget骨肉瘤),主要發生於變形性骨炎的老年患者,伴多骨損害時骨肉瘤應與轉移性骨腫瘤鑑別,約1%的變形性骨炎合併骨肉瘤但本病患者發生骨肉瘤的幾率為正常人的數千倍以上,其發病機制未明有人認為,與染色體18q21-22體質性雜合子丟失有關等發現。在染色體18q的18S60和18S42之間含有腫瘤抑制位點。丟失這些位點可導致骨肉瘤的發生,同時這一部位也是家族性變形性骨炎和家族性擴張性骨溶解(familialexpansileosteolysisFEO)的基因位點故變形性骨炎易並發肉瘤。變形性骨炎的病因存在不均一性,除18q21-22,還存在其他的遺傳位點。
疾病診斷
 變形性骨炎--診斷圖
變形性骨炎--診斷圖1、診斷
本病可與甲狀旁腺功能亢進,骨轉移瘤(特別是來自前列腺癌和乳腺癌),多發性骨髓瘤,纖維性骨發育不良相混淆,要注意鑑別常因為其他原因行X線和實驗室檢查時意外地發現本病。病骨的X線檢查特徵為密度增高,結構異常,皮質增厚,彎曲與過度生長,脛骨或股骨可見微骨折。實驗室檢查結果為血清鹼性磷酸酶升高,尿排泄羥脯氨酸總量增加,血清鈣,磷含量一般正常。骨放射核素掃描(鎝標記磷酸鹽)顯示Paget病變局部對核素的攝取量增加。
2、鑑別診斷
多數變形性骨炎患者常無臨床症狀,故在疾病的中、早期診斷較為困難。診斷中對下列臨床症狀者應懷疑變形性骨炎並應做進一步檢查:①頭顱逐年增大,伴有耳聾或其他顱神經受損症狀;②上肢或下肢出現進行性加重的弓狀畸形;③原因不明的病理性骨折;④不明原因的血ALP增高。
X線檢查有助於診斷受累及病灶區。骨端受累、溶骨區界限銳利、楔形透光區、廣泛性硬化、骨體積增大、骨小粱變粗等有助於與其他疾病鑑別。廣泛的骨密度增加應與骨轉移癌(尤其是前列腺癌骨轉移)、骨髓纖維化、腎性骨病、氟骨症、纖維異常增殖症和結節性硬化症鑑別。變形性骨炎累及顱骨時可出現顱骨肥大,應與額骨內板肥厚症、纖維異常增殖症、貧血和骨轉移癌等鑑別。本病盆骨硬化呈非對稱性或單側分布、受累骨增大、骨小粱增粗。累及脊椎時,病變椎體呈框架征,四周濃密。血管瘤所致者表現為縱向骨小粱增粗,腎性骨病除有腎臟本身的疾病外。特徵為橄欖球衣狀脊椎。
疾病檢查
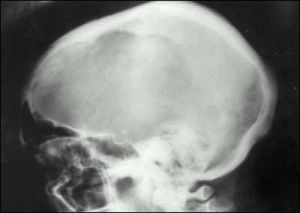 變形性骨炎--CT
變形性骨炎--CT血ALP升高有助於本病勱的診斷但正常時不能排除其可能。部分病人血鈣升高,血磷稍低。血中骨源性鹼性磷酸酶水平和尿羥脯氨酸增加。血鈣、磷、鎂和PTH一般正常。血鹼性磷酸酶(ALP)血ALP水平與病變範圍和病變的活動程度有關體積小的骨骼病變(約為10%左右)ALP正常顱骨病變時ALP升高。如並發骨肉瘤ALP可急劇增高,酸性磷酸酶和5-核苷酶也可升高。尿羥脯氨酸(HYP)正常人在低明膠飲食時的尿羥脯氨酸的排泄量低於50mg/d,而變形性骨炎患者因其骨重建旺盛,尿羥脯氨酸排泄量可高達2000mg/d。此外,尿羥賴氨酸也能反映骨重建活動的水平和本病的病變程度。血鈣、磷、鎂和PTH健康搜尋一般正常。約15%~20%的患者因骨重建對鈣的需求增加血鈣廓清加速導致血PTH上升。骨受侵部位廣泛的患者或合併原發性甲旁亢時有高血鈣症和高尿鈣症。
2、其它輔助檢查:
X線的表現較複雜可歸納為下列數點:①骨質破壞骨小梁粗糙稀疏伴局限性骨質疏鬆晚期的骨皮質與髓質腔界限不清,結構模糊如網狀;②骨幹增粗,膨大,彎曲變形,呈腰刀狀;③顱骨局限性骨質疏鬆伴棉絮狀增生,內外板界限消失,顱縫模糊,頭顱增大;④椎體呈柵欄狀和方框狀改變;⑤長骨溶骨性病灶有時呈V形;⑥骨盆連緣和弓狀線增厚,出現邊緣征;⑦髖關節間隙變窄,骨質增生短骨增粗;⑧病變區病理性骨折。
破骨細胞和成骨細胞活動均增強,破骨細胞內可找到核內包涵體硬化期的骨組織像缺乏特異性,但可發現新舊骨質混雜的鑲嵌徵象和破骨細胞的特徵性改變。
本病常伴有各種骨腫瘤、如骨肉瘤、纖維肉瘤、軟骨肉瘤、網織細胞肉瘤、巨細胞瘤和多發性骨髓瘤等,可能與染色體18q21~22的雜合子丟失有關此部位的腫瘤抑制性位點丟失和家族性擴張性肌溶解(familialexpansileosteolysis)基因激活可導致變形性骨炎並肉瘤的發生。此外,亦可與原發性甲旁亢並存。
疾病治療
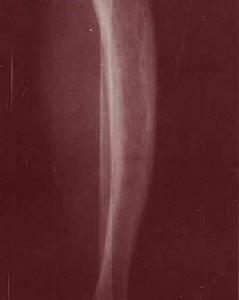 變形性骨炎影像
變形性骨炎影像1、藥物治療
(1)、降鈣素:用量較一般大,開始每天皮下或肌注鮭魚降鈣素100U(40mg),數周后改為隔天100U。骨
病基本消失後逐漸減至每周100-200U。療程至少1年,有時需長期套用。
(2)、磷酸鹽:EHDP(disodiumethane-1hydroxyl-1,1-diphosphate;disodiumetidronate),口服20mg/(kg?d),用藥時間依病情而定,一般為0.5~1年。磷酸鹽宜與降鈣素合用。
(3)、普卡黴素:具有降低血鈣,抑制骨代謝作用。靜滴15-25μg/(kg?d),連用7-10天,無明顯副作用者可酌情再繼續套用,亦可以較小劑量連用數周,或用較大劑量每1-2周靜滴1次。本藥的主要副作用是消化道反應,一過性肝腎損害及骨髓抑制等。
(4)、氟化鈉:作為輔助治療,一般與維生素D等合用。藥物治療能影響無機離子的流量,並抑制骨細胞的活性,故有治療作用。適宜化療的指征為:(1)疼痛必須明確與Paget病有關,而不是其他疾病(如骨關節炎)引起者。(2)在做矯形外科手術之前,先進行藥物治療,以防止或減少術中出血。(3)為了預防併發症的發生,或推遲其發展(如椎骨Paget病手術效果差的患者發生的下肢輕癱或截癱)。
常用的雙磷酸鈉藥物:羥乙二磷酸二鈉,5-10mg/kg口服,每日1次,療程6個月,需要時隔3-6個月後重複給藥;明顯活動期需用大劑量,每日20mg/kg,口服3個月。阿侖特羅,單用時每日40mg,口服6個月,晨起進餐前至少30分鐘服藥。氨羥二磷酸二鈉(pamidronate),每日30-90mg,靜脈輸注維持4小時,連續3天(明顯活動期劑量更大)。Tiludronate,每日400mg口服,療程為3個月。合成鮭降鈣素,每日50-100IU(0.25~0.5ml),皮下或肌內注射,開始有治療效果後(常在治療1月後)減量為50IU,隔天1次,甚至可減少至每周2次或1次。合成人降鈣素每日0.5mg,皮下注射,一個月後減量為隔日1次至每周2次。降鈣素也可通過鼻噴的方式給藥,用於接受無痛性治療的患者。
(5)、其他藥物:包括鈣劑、維生素D、氫氧化鋁、胰高糖素、放射菌素D和吲哚美辛(消炎痛)等,但療效均未肯定。
2、手術治療
顱底陷入症可考慮枕下開顱減壓。有交通性腦積水時可行腦室-頸靜脈分流術,椎板減壓和椎孔成形術可解除脊髓壓迫或神經根壓迫症狀。長骨骨折應作相應處理,有畸形者可行截骨手術等3、預後:伴發肉瘤患者有骨痛、腫脹和病理性骨折,預後很差,目前尚無有效治療的藥物,化療和手術僅能控制症狀,而對病變本身無明顯療效。放射治療和截肢可減輕疼痛。術後5年的存活率為5%-8%。
4、預防:Gold等對2000例變形性骨炎患者進行了健康自我評定(self-ratedhealth,SRH)的現狀進行調查,並與綜合性的SRH研究結果進行相關性分析。結果發現,調查的總體回歸模型與綜合研究結果相關,而病人調查結果認為,家庭對病人的幫助滿意程度、活動功能的局限、病情的壓力和憂慮症狀有著潛在的可調節性,有助於提高患者的健康感知程度,有利於提高生活質量和延長壽命。

