概述
 悉心呵護
悉心呵護1.果糖激酶缺乏症(或稱特發性果糖尿症,essentialfructosuria)是由於肝臟缺乏果糖激酶所造成,使果糖不能進行磷酸化而不能在肝臟中進一步代謝,因此患者血液中的果糖濃度在攝食果糖後明顯升高並自尿液中排出,本病無明顯臨床症狀,正確診斷的意義在於防止誤診糖尿病而妄加治療。
2.遺傳性果糖不耐症(hereditaryfructoseintolerance)系因果糖二磷酸醛縮酶(fruetaldolase,fructose-,6-diphosphatealdolase)缺陷所致是本文敘述重點。
3.果糖-1,6-二磷酸酶缺乏症(fructose-1,6-diphosphatasedeficiency)這是葡萄糖代謝途徑中的催化酶,但習慣上歸納在果糖代謝缺陷中。
流行病學
遺傳性果糖不耐症系半乳糖血症中的一種,發病率各國不同,是半乳糖分解代謝中,果糖二磷酸醛縮酶缺陷所致的代謝紊亂性疾病,其中半乳糖-1-磷酸尿苷轉移酶缺陷發病率約1∶5萬~1∶1萬果糖激酶缺陷是一種罕見和無症狀的常染色體隱性遺傳病根據在2.9萬例尿糖陽性病人中發現4例該酶缺陷者,推測其發病率為1/13萬。
病因
本病屬於常染色體隱性遺傳病以14C標記的果糖檢查病人肝果糖-1-磷酸醛縮酶,發現為正常的0~12%,呈顯著性降低。
發病機制
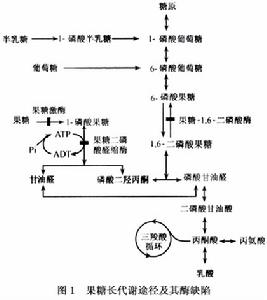 遺傳性果糖不耐受
遺傳性果糖不耐受果糖廣泛存在於各種水果和蔬菜中含量最高者可達乾重的40%並常被用作食品中的添加劑,因此人體自日常飲食中攝入的果糖量較大。果糖進入人體後大部分在肝臟中進行代謝僅小量由腎小管和小腸代謝果糖作為人體攝入的另一種單糖其體內代謝過程為:首先在肝腎及腸黏膜果糖激酶的催化下,轉變為1-磷酸果糖;後者在果糖-1-磷酸醛縮酶的作用下分解成磷酸二羥酮及甘油醛;後者通過甘油醛激酶磷酸化作用轉變成磷酸甘油醛再經果糖-1,6-磷酸醛縮酶的催化磷酸甘油醛與磷酸二羥丙酮縮合成1,6-二磷酸果糖。後者再由果糖-1,6-雙磷酸酶水解成果糖-6-磷酸和磷酸進一步轉變成6-磷酸葡萄糖最終轉變為葡萄糖或糖原。此外磷酸二羥丙酮還可循酵解-氧化途徑轉變成乙酞CoA從而合成脂肪果糖在上述酶的作用下最終約有50%轉化為葡萄糖,其餘則生成糖原丙酮酸、三酸甘油酯和脂肪等。臨床已發現有果糖激酶果糖-1-磷酸醛縮酶及果糖1,6-二磷酸酶的先天性缺陷參見圖1。
遺傳性果糖不耐症是由於果糖二磷酸醛縮酶缺陷所致。已知該酶的分子量為16萬,由4個亞單位組成;根據其催化活性免疫特徵和在不同組織中的分布情況又可分為AB、C三型同工酶在肝腎和小腸中以B型果糖二磷酸醛縮酶為主它的編碼基因位於9q13~q32,長約14500bp歐洲資料表明:A149p174D和N334k三種點突變是導致果糖不耐症的最主要原因本病患兒肝臟內的果糖二磷酸醛縮酶活性由完全缺如到僅為正常人的12%左右不等,由於該酶缺乏果糖代謝的兩個主要臟器肝和腎中都有果糖-1-磷酸堆積除對細胞有損害外,累積的1-磷酸果糖不僅可使果糖-1,6-二磷酸酶活性受到抑制使正常從甘油、胺基酸等轉變成葡萄糖的糖原異生作用受阻從而引起低血糖此外,過多的1-磷酸果糖還抑制了磷酸化酶的作用,阻礙糖原轉變成葡萄糖,這是導致低血糖的另一原因而且由於大量無機磷(Pi)亦同時被圍入,使血磷降低和ATP再生減少。1-磷酸果糖的累積和ATP供應不足兩者也阻礙了糖原釋出1-磷酸葡萄糖如繼續使用含蔗糖或果糖的食物餵養,將造成患兒肝細胞損傷,持久的含果糖飲食會造成患兒肝細胞壞死、脂肪浸潤、膽小管增生和纖維化,甚至肝硬化其機制不甚清楚,可能是由於1-磷酸果糖的細胞毒性作用或與缺乏ATP有關。
症狀
 著名醫師
著名醫師 遺傳性果糖不耐受
遺傳性果糖不耐受診斷:確診可依據:1.臨床特點從食物中摒除果糖後,臨床症狀於幾小時內消失。2.果糖耐量試驗(fructosetolerancetest)顯示血葡萄糖及血磷急速下降同時果糖脂肪酸及乳酸上升但本試驗易引起低血糖發作故宜慎用,而且所用果糖量宜減少,口服按0.5g/kg靜脈量減半兒童也可按3g/m2口服。3.活檢肝腸黏膜中酶的測定顯示酶活性顯著減低
鑑別診斷:本病應注意與幽門梗阻胃腸炎、敗血症嚴重肝炎及其他糖代謝缺陷病相鑑別。
檢查
 治療設備
治療設備2.尿液生化檢查對疑似的急症患兒都應檢測尿液果糖。持續進食果糖的患兒常有腎小管酸中毒和Fanconi綜合徵樣的腎小管再吸收障礙因此應對尿液pH蛋白胺基酸和重碳酸鹽等進行檢測。
3.果糖耐量試驗一次給予果糖200~250mg/kg靜脈快速注射後檢測血液中果糖葡萄糖無機磷、尿酸和轉氨酶可供診斷。本試驗應在病情穩定後數周進行。
4.酶學檢查可採用肝、腎或腸黏膜活檢組織進行,但非診斷必需。
治療
 葉酸保健品
葉酸保健品1.飲食療法一旦確定診斷,立即終止一切含果糖和蔗糖食物。必須終身不再攝入果糖及蔗糖由於飲食的限制維生素C攝入量減少宜予補充。
2.對症治療當出現低血糖時,靜脈內注射葡萄糖即可緩解糾正電解質紊亂,有出血傾向者可給予成分輸血。
3.支持治療對急性肝功能衰竭患兒應予以積極支持治療。
4.葉酸治療也有報告用大量的葉酸似乎是有希望的治療方法。
預防
預後:合適的治療可使所有症狀在2~3天內消失血液生化改變在2周內恢復正常生長落後情況則需2~3年始見好轉。無果糖飲食可使患兒獲得完全正常的生活但多數仍留有輕度肝腫大和脂肪變性。
預防:發病機制尚未完全清楚,參考遺傳性疾病預防措施由於在同一家系中還可出現其他病兒,因此對發病家庭的新生嬰兒要嚴密觀察。
罕見病詞條庫

世界衛生組織確認的罕見病
世界罕見病名錄
| 罕見病,為患病人數占總人口的0.65%~1%的疾病,許多罕見病的病因在醫學上並不十分明確,所以罕見病的治療是醫學上的一大難題,本任務盤點一下一些罕見病。 |
兒科疾病
| 兒科病就是兒童易患疾病的統稱。比如腸胃疾病,營養不良,呼吸疾病等等。 |
