簡介
侏儒綜合徵(Laron syndrome),又稱萊倫氏綜合徵,又稱生長激素遲鈍症候群。病因
兒童生長遲緩的情況,最常見的是生長激素不足所造成的。人類生長激素由腦下垂體分泌後,須有「受體」結合,活化特定的蛋白(月每),才能將訊息傳遞而發揮功能;同時,生長激素會刺激肝臟分泌「類胰島素生長因子」(IGF-1),執行促進生長及再生的功能。萊倫氏症候群(Laron syndrome)是一種生長嚴重遲緩的罕見疾病,也稱為「生長激素遲鈍症候群」。此症臨床上可分為兩型,第一型是由於生長激素的受體基
 侏儒症
侏儒症症狀
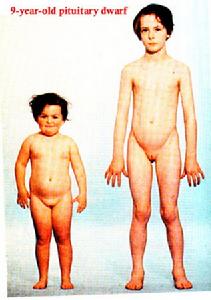
萊倫氏症候群的病人在臨床表現上有嚴重的生長遲緩,迷你的身材是同齡者等比例縮小,搭配上藍色的鞏膜,看起來就像是洋娃娃,他們不同於軟骨不全症患者的頭臉身體正常、四肢短小,也與透納氏患者窄臉、短頸不一樣。其他的症狀有:骨齡延遲、馬鞍鼻、聲音音調高亢、男性生殖器較小、骨質疏鬆、肌肉發育不良、運動能力發展遲緩等症狀。從生化檢查可以發現有低血糖(嬰兒期)及胰島素低下的情形;血漿中的生長激素通常會升高,但相同的病人數值差距卻非常的大;類胰島素生長因子-1的量會降低,和一般生長激素缺乏的患者不同。另外我們檢測生長激素結合蛋白,可以發現第一型的患者數值較低,而第二型則正常。
診斷
 侏儒症的治療
侏儒症的治療治療
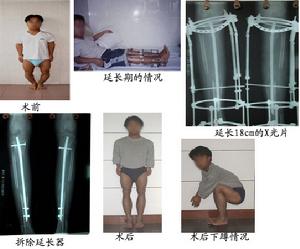
目前臨床醫學上,使用一種人類基因重組的類胰島素生長因子-1(recombinant human IGF-1)來治療,普遍來看有不錯的療效。因為它可以刺激成骨細胞生長,能增加骨質密度,刺激肌肉細胞生長和分化,防止肌肉蛋白質的降解,增加肌肉組織含量,並具有促進血液流通和細胞再生、提昇新陳代謝、增加人體整體體能的功能。對於嬰兒照護方面,要注意多加以餵食,監測血糖以避免低血糖的情況發生。罕見病詞條庫

盤點常見的遺傳病
| 隨著分子生物學的發展,科學家們成功的把基因和疾病聯繫在一起。人們可以預先通過基因篩查來預測可能罹患的遺傳病。比如,GOOGLE公司創始人之一謝爾蓋?布林,就在其妻子的23andme生物技術公司通過測序發現自己可能在未來患帕金森氏綜合症,他積極通過各種方式來預防疾病的發生並投入巨資進行相關的研究。但如同一把雙刃劍,基因篩查也使一些攜帶突變基因的人在社會各個方面受到歧視,讓我們來關注一下遺傳疾病吧! |
| 黑棘皮症| 短趾症| 家族性高膽固醇血症| 白化病| 苯丙酮尿症| 紅綠色盲| 抗維生素D佝僂病| 先天性髖關節脫位| 脊柱裂| 先天性聾啞| 蠶豆病 | 強直性肌營養不良| 亨廷頓氏病| 老年性痴呆症| 精神分裂症| 脆性X綜合徵 | 原發性高血壓| WILSON 氏病| 成人多囊腎病| 馬凡綜合徵 | 侏儒綜合徵| 視網膜色素變性| 糖原貯積症| 神經鞘脂貯積症| 黏多糖貯積症 | 半乳糖血症| 白癜風| 性反轉綜合徵| WILMS 瘤| 視網膜母細胞瘤| 成骨不全病| 自毀容貌症| 家族性多發性結腸息肉病| 強直性脊柱炎| 假肥大性肌營養不良| 牛皮癬| 多囊腎| 脆骨病| 神經纖維瘤| 上瞼下垂| 類風濕性關節炎| 癲癇| 先天性心臟病| 高膽固醇血症| 唇裂| 齶裂| 魚鱗症| 多指| 著色性乾皮病| 腓肌萎縮症| 並指| 畸形足| 青光眼| 全身自化| 結腸息肉症| 先天聾啞| 原發性小睪症 |
