
簡介
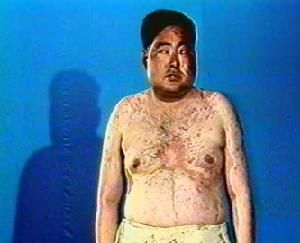 庫欣綜合徵
庫欣綜合徵庫欣綜合徵是指凡由於體內皮質醇過多而產生的臨床症候群,過去稱此病症為“柯興氏綜合徵”,把由於垂體分泌過量促腎上腺皮質激素(ACTH)而引起的腎上腺皮質增生症稱為“柯興氏病”。伊森科(Иценко)在1925年曾提出此病症在垂體和間腦有病變的觀點,故亦稱之為“伊森科-柯興氏綜合徵”。
庫欣(曾稱皮質醇增多症)綜合徵,是腎上腺皮質功能亢進症中的最常見的一種。高血壓為庫欣綜合徵的常見表現,約90%的病人有血壓升高,一般在150/100毫米汞柱。
特點
庫欣綜合徵主要由於腎上腺皮質分泌過多的皮質醇所引起的臨床一系列症狀、體徵,但也常分泌其它激素,所以症群多變異,屬於皮質醇為主的混合型。本病的主要特點:
1)脂肪代謝紊亂,使病人面如滿月(滿月臉)、紅潤、多脂、毛髮油膩、臉頸及軀幹肥胖(向心性肥胖)、水牛背、鬚毛增多等。
2)糖代謝紊亂,使病人在臨床上出現了糖尿病症群和糖尿,約占本病的10%~30% ,稱為類固醇性糖尿病。
3)蛋白質代謝紊亂,臨床上出現蛋白質過度消耗現象,如皮膚菲薄、毛細血管脆性增加易發生淤斑和紫紋並呈對稱性,骨質疏鬆及抵抗力低下易發生痤瘡等。
高血壓為庫欣綜合徵的常見表現,約90%的病人有血壓升高,一般在150/100毫米汞柱。病人主訴頭痛、頭暈、胸悶、心悸、視力模糊等症狀。長期高血壓可並發左心室肥厚、心肌勞損、心律失常、心力衰竭、腦血管意外和腎功能衰竭,有時有蛋白尿和低滲尿等。
類型
 庫欣綜合徵
庫欣綜合徵醫源性皮質醇症
長期大量使用糖皮質激素治療某些疾病可出現皮質醇症的臨床表現,這在臨床上十分常見。這是由外源性激素造成的,停藥後可逐漸復原。但長期大量套用糖皮質激素可反饋抑制垂體分泌ACTH,造成腎上腺皮質萎縮,一旦急驟停藥,可導致一系列皮質功能不足的表現,甚至發生危象,故應予注意。長期使用ACTH也可出現皮質醇症。
垂體性雙側腎上腺皮質增生
雙側腎上腺皮質增生是由於垂體分泌ACTH過多引起。其原因:①垂體腫瘤。多見嗜鹼細胞瘤,也可見於嫌色細胞瘤;②垂體無明顯腫瘤,但分泌ACTH增多。一般認為是由於下丘腦分泌過量促腎上腺皮質激素釋放因子(CRF)所致。臨床上能查到垂體有腫瘤的僅占10%左右。這類病例由於垂體分泌ACTH已達一反常的高水平,血漿皮質醇的增高不足以引起正常的反饋抑制,但口服大劑量氟美松仍可有抑制作用。
垂體外病變引起的雙側腎上腺皮質增生
支氣管肺癌(尤其是燕麥細胞癌)、甲狀腺癌、胸腺癌、鼻咽癌及起源於神經嵴組織的腫瘤有時可分泌一種類似ACTH的物質,具有類似ACTH的生物效應,從而引起雙側腎上腺皮質增生,故稱異源性ACTH綜合徵。這類患者還常有明顯的肌萎縮和低血鉀症。病灶分泌ACTH類物質是自主的,口服大劑量氟美松無抑制作用。病灶切除或治癒後,病症即漸可消退。
腎上腺皮質腫瘤
大多為良性的腎上腺皮質腺瘤,少數為惡性的腺癌。腫瘤的生長和分泌腎上腺皮質激素是自主性的,不受ACTH的控制。由於腫瘤分泌了大量的皮質激素,反饋抑制了垂體的分泌功能,使血漿ACTH濃度降低,從而使非腫瘤部分的正常腎上腺皮質明顯萎縮。此類患者無論是給予ACTH興奮或大劑量氟美松抑制,皮質醇的分泌量不會改變。腎上腺皮質腫瘤尤其是惡性腫瘤時,尿中17酮類固醇常有顯著增高。
腎上腺皮質腫瘤多為單個良性腺瘤,直徑一般小於3~4cm,色棕黃,有完整的包膜。瘤細胞形態和排列與腎上腺皮質細胞相似。腺癌則常較大,魚肉狀,有浸潤或蔓延到周圍臟器,常有淋巴結和遠處轉移。細胞呈惡性細胞特徵。 無內分泌功能的腎上腺皮質腫瘤則不導致皮質醇症。
臨床上發現少數病例腎上腺呈結節狀增生,屬增生與腺瘤的中間型。患者血漿ACTH可呈降低,大劑量氟美松無抑制作用。
臨床上70%的病例為垂體病變所致的雙側腎上腺皮質增生,良性腺瘤占20~30%,惡性腎上腺腺癌占5~10%,異位ACTH分泌過多則甚為少見。
臨床表現
典型的庫欣綜合徵的臨床表現主要是由於皮質醇分泌的長期過多引起蛋白質、脂肪、糖、電解質代謝的嚴重紊亂及干擾了多種其他激素的分泌。此外,ACTH分泌過多及其他腎上腺皮質激素的過量分泌也會引起相應的臨床表現。包括:向心型肥胖,蛋白質過度消耗現象,糖代謝紊亂,電解質紊亂,心血管變化,免疫力降低,造血及血液系統變化,性功能障礙,神經精神障礙,皮膚色素沉著。
檢查
1.X線檢查
(1)蝶鞍平片法或分層攝片法由於庫欣病患者的垂體腫瘤較小,平片法結果大多陰性,用蝶鞍分層片法部分病人僅有輕度的異常改變,且敏感度差,準確性不大。但如發現蝶鞍增大,有助於垂體瘤的診斷。
(2)腎上腺X線法對腎上腺占位性病變的定位有幫助,但不能鑑別結節性增生與腺瘤。
2.CT檢查
由於CT掃描的每1層約10mm,對於直徑>10mm的垂體腺瘤,CT的解析度良好,但對直徑<10mm的垂體微腺瘤,CT有可能遺漏,陽性率可達60%。所以CT。未發現垂體瘤者,不能排除微腺瘤的可能。
對腎上腺增生與腺瘤的檢查,CT的作用大,解析度好,因為腎上腺腺瘤的直逕往往>2cm。
注意:CT檢查,要注射造影劑,為了防止變態反應,一般都給予10mg地塞米松;CT檢查要安排在大劑量的地塞米松抑制試驗以後,否則要間隔7天以上再做大劑量的地塞米松抑制試驗。
3.磁共振(MRI)檢查
對庫欣病,MRI是首選方法,與CT相比可較好地分辨下丘腦垂體及鞍旁結構(海綿竇、垂體柄和視交叉),但對直徑<5mm的腫瘤,解析度仍僅為50%。
4.B超
對腎上腺增生與腺瘤好,屬無創傷檢查,方便、價廉、較準確。常用來與MRI,CT一起作庫欣綜合徵的定位診斷。
5.其他
(1)131Ⅰ-α-碘化膽固醇腎上腺掃描能顯示腎上腺腺瘤的部位和功能;腺瘤側濃集,對側往往不顯影,圖像不如CT清晰。
(2)岩下竇ACTH測定(IPSS)做選擇性靜脈取血,測ACTH。若病人經生化檢查為庫欣病,而CT等掃描為陰性,可做此檢查。
診斷
皮質醇症的診斷分三個方面:確定疾病診斷、病因診斷和定位診斷。
1.確定疾病診斷
主要依典型的臨床症狀和體徵。如向心性肥胖、紫紋、毛髮增多、性功能障礙、疲乏等。加上尿17羥皮質類固醇排出量顯著增高,小劑量氟美松抑制試驗不能被抑制和血11羥皮質類固醇高於正常水平並失去晝夜變化節律即可確診為皮質醇症。早期輕型的病例應與單純性肥胖相鑑別。
2.病因診斷
即區別是由腎上腺皮質腺瘤、腺癌、垂體腫瘤引起的皮質增生、非垂體腫瘤或異源性ACTH分泌腫瘤引起的皮質增生。
3.定位診斷
主要是腎上腺皮質腫瘤的定位,以利手術切除。但定位的同時,也常解決了病因診斷。
治療
手術療法
 庫欣綜合徵
庫欣綜合徵2、腎上腺皮質腫瘤摘除 適用於腎上腺皮質腺瘤及腎上腺皮質腺癌。如能明確定位,可經患側第11肋間切口進行。如不能明確定位,則需經腹部或背部切口探查雙側腎上腺。腎上腺皮質腺瘤摘除術較簡單,但腎上腺皮質腺癌者常不能達到根治。由於腫瘤以外的正常腎上腺呈萎縮狀態,故術前、術後均應補充皮質激素。術後尚可肌注ACTH20r/d,共2周,以促進萎縮的皮質功能恢復。術後激素的維持需達3個月以上,然後再逐步減量至停服。
3、雙側腎上腺摘除 適用於雙側腎上腺皮質增生病例。其方法有①雙側腎上腺全切除:優點是控制病情迅速,並可避免復發;缺點是術後要終身補充皮質激素,術後易發生Nelson症(垂體腫瘤+色素沉著)。②一側腎上腺全切除,另一側腎上腺次全切除:由於右側腎上腺緊貼下腔靜脈,如有殘留腎上腺增生復發,再次手術十分困難,故一般作右側腎上腺全切除。左側殘留腎上腺應占全部腎上腺重量的5%左右。殘留過多,則復發率高。殘留過少或殘留腎上腺組織血供損傷,則出現腎上腺皮質功能不全或Nelson症。故術中應注意勿損傷其血供。由於腎上腺血供是呈梳狀通向其邊緣,故殘留的組織應是邊緣的一小片組織。有的作者採用一側腎上腺全切除加垂體放療,但常無效或有復發。
在作腎上腺手術時,應注意以下幾點:①切口的選擇:可經第11肋間切口進行,但術中需更換體位,部分腎上腺皮質腺瘤病人誤診為腎上腺皮質增生時,則發生困難。病人肥胖,經腹部探查雙側腎上腺較困難。比較合適的是病人全麻下取俯臥位,經背部八字切口(Nagamatsu切口(圖1),或經第11肋切口探查。一般先探查右側,如發現右側腎上腺增生(雙側腎上腺增生)或萎縮(左側腎上腺皮質腺瘤),則需再探查左側腎上腺。如發現右側腎上腺皮質腺瘤則可作腺瘤摘除,不需再探查左側。巨大的腎上腺腺癌可選用胸腹連合切口進行手術(圖2)。②皮質激素的補充:皮質醇症患者體內皮質醇分泌處於一高水平,術後皮質醇水平驟降易導致急性腎上腺皮質功能不足所致的危象。其臨床表現為休克、心率快、呼吸急促、紫紺、噁心嘔吐、腹痛、腹瀉、高熱、昏迷甚至死亡。故於術前、術中和術後均應補充皮質激素以預防。一旦危象發生,應快速靜脈補充皮質激素,糾正水電解質紊亂以及對症處理。情緒波動、感染以及某些手術併發症可誘發危象發生,並有時會混淆診斷(如氣胸、出血等),應予注意避免發生。
以上補充的皮質激素量雖已超過正常生理分泌量,但由於術前患者皮質醇分泌處於一很高水平,故部分病例仍可發生危象。由於術後危象大多發生於手術後2天之內,故我院於術日及術後2天再靜脈補充氫化可的松100~200mg/d, 從而使危象的發生大大減少。如疑有危象或有手術併發症,均應加大皮質激素用量。皮質激素的長期維持量是醋酸可的松25~37.5mg/d(為正常生理需要量)。腺瘤患者一般需維持3~6個月後停藥,雙側腎上腺全切除者需終生服藥。如病人有其它疾病、感染及拔牙等手術時,應增大激素用量。如有腹瀉及不能進食時,應改成肌注用藥。病人應隨身攜帶診斷書,隨時供醫生參考。腎上腺腺瘤及腎上腺大部切除患者在病情穩定後可逐步停藥。停藥前如需測定體內皮質醇分泌水平,可停服醋酸可的松,改服氟美松(0.75mg氟美松相當於25mg醋酸可的松)1~2周,再測24小時尿17羥、17酮的排出量。因氟美松不影響尿中17羥、17酮類固醇的測定,故所測得的17羥、17酮類固醇表示體內皮質醇的分泌水平。如已接近正常,則可逐步減量停藥。如水平極低,則仍繼續改服醋酸可的松維持。有作者報導將切除的腎上腺切成小塊,埋植在縫匠肌或腸系膜中治療手術後腎上腺皮質功能低下,獲得一定療效。經放射性核素標記膽固醇掃描證明移植區確有放射性濃集,尿17-羥類固醇排出量也有升高,部分病例可停服或減少皮質激素的維持量。如有皮質功能亢進者,可局部作一較小手術切除之。由於腎上腺動脈細小,帶血管的自體腎上腺移植有一定困難。③Nelson症的處理:腎上腺全切除後,垂體原有的腺瘤或微腺瘤可繼續增大,壓迫視神經,引起視力障礙。垂體分泌的促黑色素激素引起全身皮膚黏膜色素沉著,甚至呈古銅色。垂體腺瘤摘除術可以挽救視力,垂體局部放療可以抑制腫瘤的生長。中醫中藥對緩解色素沉著也有一定療效。
非手術療法
1、垂體放射治療 有20%病例可獲持久療效。但大多數病例療效差且易復發,故一般不作首選。垂體放療前必須確定腎上腺無腫瘤。
2、藥物治療 副作用大,療效不肯定。主要適用於無法切除的腎上腺皮質腺癌病例。①二氯二苯二氯乙烷(O,P´DDD,dichlorodiphenyldichloroethane):可使腎上腺皮質網狀帶和束狀帶細胞壞死。適用於已轉移和無法根治的功能性或無功能性的皮質癌。但有嚴重的胃腸道和神經系統的副作用,並可導致急性腎上腺皮質功能不足。治療劑量4~12g/d,從小劑量開始漸增到維持量,並根據病人忍受力和皮質功能情況調節。②甲吡酮(metyrapone,Su4885):是11β-羥化酶抑制劑。可抑制11-去氧皮質醇轉化為皮質醇、11-去氧皮質酮轉化為皮質酮,從而使皮質醇合成減少。副作用小,主要為消化道反應。但作用暫時,只能起緩解症狀的作用。一旦皮質醇分泌減少刺激ACTH的分泌,可克服其阻斷作用。③氨基導眠能(aminoglutethimide):可抑制膽固醇合成孕烯醇酮。輕型腎上腺皮質增生症服1~1.5g/d,嚴重者1.5~2g/d可控制症狀。但需密切隨訪皮質激素水平,必要時應補充小劑量的糖皮質激素和鹽皮質激素,以免發生腎上腺皮質功能不足現象。④賽庚啶(cyproheptadine):是血清素(serotonin)的競爭劑,而血清素可興奮丘腦-垂體軸而釋放ACTH,故賽庚啶可抑制垂體分泌ACTH。適用於雙側腎上腺增生病例的治療。劑量由8mg/d逐漸增加到24mg/d。在雙側腎上腺全切除或次全切除術後皮質功能不足的情況下,一方面補充皮質激素,一方面服用賽庚啶能減少垂體瘤的發生機會。其它尚報告溴隱亭、腈環氧雄烷(trilostane)等藥物亦有一定療效。
鑑別
一、單純性肥胖及2型糖尿病:可有肥胖、高血壓、糖代謝異常、月經紊亂、皮膚白紋等,血尿皮質醇及其代謝產物增高,但可被小劑量地塞米松所抑制,皮質醇及ACTH節律正常。
二、假性Cushing綜合徵:酒精性肝臟損害時,不僅各種症狀及激素水平類似本病,且對小劑量地塞米松給藥無反應或反應減弱,但戒酒即可恢復。
三、抑鬱症:雖增高的激素及其代謝物不受地塞米松小劑量給藥抑制,但無Chushing綜合徵的臨床表現。
預防
病情觀察1、肥胖狀態,高血壓。
2、皮膚乾燥、皮下出血、痤瘡、創傷化膿、四肢末梢紫紺、水腫、多毛、肌力低下、乏力、疲勞感、骨質疏鬆與病理性骨折等。
3、尿量,尿性狀血尿、蛋白尿、尿糖。
4、精神症狀失眠、不安、抑鬱、興奮。
5、感染症狀發熱。
6、女性患者月經異常等。
對症護理
1、預防感染,保持皮膚清潔,勤沐浴,換衣褲,保持床單位的平整清潔。做好口腔、會陰護理。2、觀察精神症狀與防止發生事故。患者煩躁不安,異常興奮或抑鬱狀態時,要注意嚴加看護,防止墜床,用床檔或用約束帶保護患者,不宜在患者身邊放置危險品,避免刺激性言行,耐心仔細,應多關心照顧。
3、腎上腺癌化療的患者觀察有無噁心、嘔吐、嗜睡、運動失調和記憶減退。
4、每周測量身高、體重,預防脊柱突發性壓縮性骨折。
5、正確無誤做好各項試驗,及時送驗。
一般護理
1、臥床休息,輕者可適當活動。
2、飲食宜給予高蛋白、高維生素、低脂、低鈉、高鉀的食物,每餐不宜過多或過少,要均勻進餐。
健康指導
1、指導患者在日常生活中,要注意預防感染,皮膚保持清潔,防止外傷,骨折。2、指導患者正確地攝取營養平衡的飲食,給予低鈉、高鉀、高蛋白的食物。
3、遵醫囑服用藥,不擅自減藥或停藥。
4、定期門診隨訪。
飲食保健
營養飲食原則1)低鹽飲食:每日只可用3~5克食鹽。日常飲食應選擇含鈉較低的食物,如豆類及豆製品、蔬菜類、果類等。
2)進食含鉀高的食物:含鉀高的食物有:鮮香菇、黃瓜、柑橘、甜玉米、糯米、馬鈴薯、桂圓、葡萄、椰子、柿子、西瓜、芒果等。
3)多食鹼性食品:鹼性食品有:豆類、蔬菜、水果、栗子、百合、奶類、藕、蛋清、海帶、茶葉等。
4)高蛋白飲食:高蛋白食物有:黃豆、蠶豆、豌豆、花生、牛肉、豬肉、雞肉、鴨肉、內臟、雞蛋、奶粉等。
5)高維生素飲。
併發症
有研究報導此病可並發多囊卵巢綜合徵及心理障礙等疾病。
1.高尿鈣和腎結石皮質醇促進尿鈣排出,使尿鈣明顯增多,久病者可形成腎結石伴尿路結石症候群和異位鈣鹽沉積等表現。
2.高血壓和低血鉀長期高血壓可導致左心衰竭,腦動脈硬化,腦卒中等。
3.少年兒童時期發病的庫欣綜合徵患者,生長停滯,青春期遲延。

