
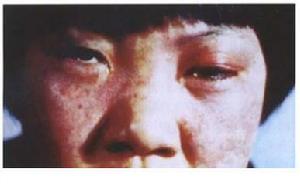
 腦三叉神經血管瘤病
腦三叉神經血管瘤病三叉神經血管瘤病(encephalotrigeminal angiomatosis)首先為Schiremer所描述以後SturgeWeber相繼作了詳細的報導,故名Sturge-Weber綜合徵。本徵是惟一無遺傳傾向的斑痣性錯構瘤病(phacoma-toses),是一種頭面部血管畸形的發育性疾病特徵是三叉神經分布的顏面區域有皮膚、黏膜毛細血管瘤,有時合併顱內血管瘤或侵犯眼部。
病因
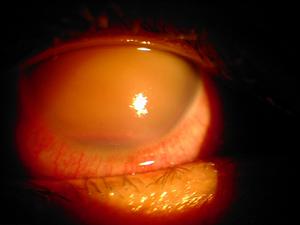 青光眼
青光眼發病機制:
青光眼的發病機制尚未完全明白。多數學者認為青光眼的發生是由於虹膜角膜角異常,如鞏膜突發育不良葡萄膜小梁網變厚、周邊虹膜向前止於小梁網上等引起。有人認為Sturge-Weber綜合徵是因為房水分泌過多及葡萄膜血管的滲透性增加所致。一些病理標本提示有虹膜角膜角結構的異常,與原發性先天性青光眼相似,阻礙了房水的外流和排出。近期的研究認為,多數病例的眼壓升高是由於淺層鞏膜靜脈壓的升高致使房水流暢係數降低所致。而淺層鞏膜靜脈壓的升高則是由於Schlemm管附近的集合管道(collector channels)呈高壓狀態所致。嬰幼兒期眼壓升高合併眼球擴張率約占70%。亦有到青年甚至晚年才發生類似原發開角型青光眼的表現。
多數臨床資料表明血管壓迫三叉神經根是原發性三叉神經痛的主要病因。血管壓迫學說認為血管壓迫與三叉神經痛之間有肯定的關係有或無三叉神經痛的同齡人血管接觸有質和量的區別所謂壓迫指血管在神經根上形成壓跡或引起神經根扭曲變形。三叉神經痛、面肌痙攣和舌咽神經痛是由於相應的腦神經在根部受到血管搏動性壓迫所致,該區對搏動性和跨過性壓迫特別敏感,而該區以外的周圍神經軸突因有施萬細胞包裹不會發生微血管壓迫,動脈粥樣硬化性動脈延長會加重這個過程。血管對神經根的壓迫,使神經纖維擠壓在一起繼而使之發生脫髓鞘變,從而引起相鄰神經纖維之間偽突觸形成,即發生“短路”。輕微的觸覺刺激即可形成一系列的衝動通過“短路”傳入中樞,而中樞的傳出衝動也可通過“短路”而成為傳入衝動,如此很快達到一定的“總和”,而引起一陣劇烈的疼痛,直至參與此過程的神經元疲憊為止經過長短不一的間歇期後,又重複上述過程。
臨床表現
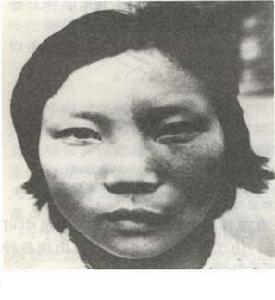 cccc
cccc2.中樞神經系統的血管瘤 在顏面部血管瘤的同側,常伴有腦膜葡萄狀血管瘤,由位於蛛網膜下擴張的靜脈組成,常累及大腦的枕葉及顳葉。頭顱平片的X線檢查(CT或磁共振成像則更佳)顯示出在血管下的腦皮質常有進行性鈣化改變,呈珊瑚狀,此鈣化征多在1歲以後患兒才出現,提示中樞神經系統已受累。腦的損害常表現為癲癇大發作(占80%病例)、皮質性癲癇發作或對側輕度偏癱甚至半身不遂和同側偏盲。多於60%的病例由於鄰近大腦皮質的萎縮而有不同程度的精神障礙。
Klippel-Trcnaunary-Weber綜合徵與本綜合徵相似或許就是其中的一種類型尚有受累側軀幹及肢體有血管瘤和靜脈曲張,有骨骼和軟組織的增生肥大。
3.眼部表現 眼部受血管瘤侵犯的情況常見,如視網膜、結膜、淺層鞏膜、睫狀體及眼瞼等均可受累虹膜有異色及增生改變。
(1)脈絡膜血管瘤:約半數患者有此征為孤立、橘黃色、中度隆起的塊狀物,位於眼底後極部。如受累範圍較廣,則眼底呈瀰漫紅色,稱為“蕃茄醬”眼底(“tomato cat-sup”fundus),脈絡膜血管瘤上可見視網膜囊樣變性視網膜水腫或繼發滲出性視網膜脫離。螢光血管造影顯示脈絡膜呈大量屈曲擴張小血管形成的異常強螢光。
(2)青光眼:30%的病例伴有青光眼當血管瘤累及眼瞼或結膜尤其是上瞼時,通常同側眼有青光眼。當顏面血管瘤為單側時青光眼多為同側眼發生但也有例外。皮膚血管瘤為雙側面部者,青光眼可為單側或雙側眼。顏面血管瘤伴有脈絡膜血管瘤者,大都並發青光眼。但不伴有Sturge-Weber綜合徵的脈絡膜血管瘤患者,則很少發生青光眼。
多數青光眼在嬰兒期已發生,但到兒童及青少年期才發展如早期即發展則眼球會增大,表現與其他先天性青光眼相似,後期才發展者則角膜直徑保持正常。多數學者認為Sturge-Weber綜合徵合併的青光眼其眼前段與典型的嬰幼兒青光眼無明顯差異,通常房角結構的外觀是正常的。縱使在高眼壓狀態下,如壓迫頸靜脈或用房角鏡間歇加壓眼球也可使血液在Schlemm管內反流,房角檢查見虹膜根部有較多的血管視盤甚至在長期中度眼壓升高的情況下也可保持正常色澤。當然,如果眼壓不控制,最終視盤也會變蒼白。在眼壓測量及眼壓描記時可見到眼壓計的指針出現與心率相應的搏動,這種搏動是由於擴張的血管網被間歇通行的血液充盈所引起的。
併發症:腦膜葡萄狀血管瘤、癲癇、鼻腔內血管瘤等。
診斷:根據典型的臨床表現,並結合輔助檢查不難診斷。
檢查:X線、CT或磁共振可明確大腦血管瘤下皮質的鈣化性改變及腦萎縮改變,出現癲癇發作可行腦電圖檢查;血管瘤累及脈絡膜時可行B超檢查以確定診斷;伴發青光眼時注意監測眼壓變化趨勢。
治療
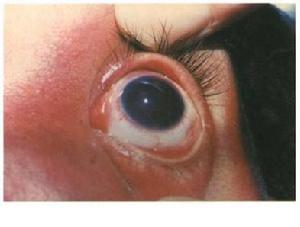 cccc
cccc濾過性手術及藥物治療均不能控制眼壓時,可試行睫狀體冷凝術,總的來說,這類青光眼對藥物或手術治療的效果往往都比較差。
預後
 微血管
微血管對復發三叉神經痛的處理,迄今無統一意見。有主張積極再手術探查,有主張改用藥物或半月節毀損治療等。應根據分析不同的可能原因、復發發生的時間、病人的年齡和全身狀態等綜合考慮。下列情況,應再次手術探查:①術後近期(3個月)發生;②不能排除手術技術因素;③病人全身情況良好,能耐受手術。再次手術探查時,除根據不同原因給予相應處理外對無明確原因者,可作三叉神經感覺根切斷術。

