概述
糖原是一種存在於細胞漿內的容易利用的儲備能源。細胞內糖原沉積(intracellularaccumulationofglycogen)發生於葡萄糖和糖原代謝異常的患者。糖原為水溶性,在非水溶性固定劑(如純酒精)中保存較好。在一般HE染色切片中,糖原被溶去呈透明的泡狀;PAS(periodicacid-schiff)染色中,呈玫瑰紅色。細胞內糖原沉積常發生於糖尿病(diabetesmellitus)患者的近曲小管遠端的上皮細胞內,甚至肝細胞、心肌細胞和胰島β細胞內。患糖原沉著病(glycogenstoragedisease)時,由於患者糖原合成或降解的酶缺陷,也導致糖原在細胞內沉積。
糖原代謝
 糖原貯積病
糖原貯積病流行病學
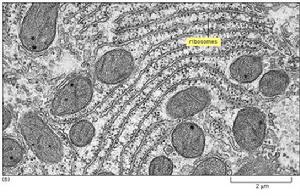 糖原貯積病
糖原貯積病病因:
糖原貯積病為常染色體隱性遺傳,磷酸化酶激酶缺乏型則是X-性連鎖遺傳 。
發病機制:
糖原貯積病系遺傳性糖原代謝紊亂。
糖原在機體的合成與分解是在一系列的酶的催化下進行的,當這些酶缺乏時, 糖原難以正常分解與合成,累及肝、腎、心、肌肉甚至全身各器官,出現肝大、低血糖、 肌無力、心力衰竭等。
臨床表現
糖原貯積病主要表現為肝大、 低血糖,包括Ⅰa型(葡萄糖-6-磷酸酶缺乏)及更罕見的Ⅰb型(G-6-P微粒體轉移酶缺乏) Ⅲ型,Ⅵ型和伴X染色體與常染色體隱性遺傳的磷酸酶b激酶缺乏。肌-能量障礙性糖原貯積病主要表現為肌肉萎縮、肌張力低下、運動障礙,包括Ⅴ型,Ⅶ型,磷酸甘油變位酶缺乏和LDHM亞單位缺乏 另有Ⅱ型、Ⅳ型等。
1.Ⅰ型糖原貯積病 臨床最常見,由於缺乏葡萄糖-6-磷酸酶,不能將6-磷酸葡萄糖水解為葡萄糖。主要表現:
(1)空腹誘發嚴重低血糖 患兒出生後即出現低血糖,驚厥以至昏迷。長期低血糖影響腦細胞發育,智力低下,多於2歲內死亡。
(2)伴酮症和乳酸性酸中毒。
(3)高脂血症,臀和四肢伸面有黃色瘤 向心性肥胖,腹部膨隆,體型呈“娃娃”狀。
(4)高尿酸血症。
(5)肝細胞和腎小管上皮細胞大量糖原沉積。新生兒期即出現肝臟腫大,腎臟增大。當成長為成人,可出現單發或多發肝腺瘤,進行性腎小球硬化、腎功能衰竭。
(6)生長遲緩,形成侏儒狀態。
2.Ⅱ型糖原貯積病 全身組織均有糖原沉積,尤其是心肌糖原浸潤肥大明顯。嬰兒型,最早於出生後1個月發病,很少生存到1歲, 面容似克汀病,舌大,嗆咳,呼吸困難,2歲前死於心肺功能衰竭, 青少年型主要表現為進行性肌營養不良。成人型表現為骨骼肌無力。
3.Ⅲ型糖原貯積病 堆積多分支糖原,又稱界限糊精病。主要表現:
(1)低血糖:較Ⅰ型輕微。
(2)肝臟大,可發展為肝纖維化,肝硬化。
(3)生長延遲。
4.Ⅳ型糖原貯積病 堆積少分支糖原,又稱支鏈澱粉病。肝大、肝硬化,生長障礙,肌張力低,如初生嬰兒有肝硬化者應除外糖原貯積病。患兒多於1周歲內死於心臟和肝臟衰竭。
5.Ⅴ型糖原貯積病 因肌肉缺乏磷酸化酶 患者肌肉中雖有高含量糖原, 但運動後血中少或無乳酸。多青少年發病,中度運動不能完成,小量肌肉活動不受限制,肌肉易疲勞,肌痙攣,有肌球蛋白尿。
6.Ⅵ型糖原貯積病 主要表現為肝大, 低血糖較輕或無。
7.Ⅶ型糖原貯積病 運動後肌肉疼痛、 痙攣,有肌球蛋白尿。輕度非球形紅細胞溶血性貧血
8.磷酸酶b激酶缺乏症(Ⅷ或Ⅸ型) 肝大,偶有空腹低血糖,生長遲緩,青春期自行緩解。
9.Ⅹ型糖原貯積病 肝臟、肌肉糖原沉積,肝臟腫大,空腹低血糖, 肌肉痙攣,一定程度智力低下。
10.O型為糖原合成酶缺乏 患者通常出現空腹低血糖,高血酮,肌肉痙攣和一定程度的智力障礙, 易與低血糖性酮症相混淆。
診斷
1.Ⅰ型診斷依據
(1)臨床表現:肝大、空腹低血糖、身材矮小、肥胖等。
(2)血液生化檢查:空腹血糖低, 血三醯甘油及膽固醇升高,血乳酸、尿酸升高。
(3)胰高糖素試驗:胰高糖素0.5mg肌內注射,每15分鐘測血糖 持續2h,正常人10~20min後空腹血糖可上升3~4mmol/L,糖原貯積病患者上升<0.1mmol/L 2h內血糖仍不升高,乳酸上升3~6mmol/L,並加重已有的乳酸性酸中毒,血pH值降低。
(4)肝穿刺活檢:是糖原貯積病確診依據 測定患者肝糖原常超過正常值6%, 葡萄糖-6-磷酸酶活性降低以至缺失,細胞核內有大量糖原沉積。
(5)果糖或半乳糖轉變為葡萄糖試驗:迅速靜脈輸注果糖(0.5g/kg)或半乳糖(1g/kg)配製的25%溶液,每10分鐘取血1次 共1h,測定血葡萄糖、乳糖、果糖、半乳糖含量,患者血葡萄糖不升高,而乳酸明顯上升。
(6)骨骼X線檢查:可見骨骺出現延遲及骨質疏鬆。
2.Ⅱ型診斷依據
(1)症狀和體徵:患兒生長發育落後,心臟肥大,肌肉鬆弛。
(2)肌酸磷酸酶和醛縮酶增高。
(3)確診依賴肌肉、肝臟活檢,電鏡示糖原顆粒沉積,缺乏α1 4-葡萄糖苷酶, 皮膚活檢成纖維細胞培養也無此酶的存在。
(4)早期妊娠時羊水細胞中可見糖原顆粒。
3.Ⅲ型診斷依據
(1)症狀和體徵:肝大、肌無力。
(2)胰高糖素試驗:清晨空腹肌內注射0.5mg後,患者血糖不升或上升很少;進食2h後肌內注射0.5mg,血糖可上升3~4mmol/L,血乳酸濃度不變。
(3)肝臟或肌肉活檢:用碘測定呈紫色反應,證實有界限糊精存在。也可作紅細胞、 白細胞加碘檢測。
(4)紅細胞、白細胞澱粉α1,6-葡萄糖苷酶活性測定。
4.Ⅳ型診斷依據
患兒有肝硬化,肝脾腫大,黃疸和腹水、 肝組織碘試驗澱粉呈紫色反應者為陽性
5.Ⅴ型診斷依據
(1)症狀和體徵:肌肉活動受限,肌痙攣等。
(2)束臂運動試驗:患者上臂扎血壓帶,打氣使氣帶壓力達收縮期血壓以阻斷血流,然後讓病人伸曲手指反覆運動1min,於運動前後測該臂血乳酸,正常人運動後乳酸增高,而患者血乳酸不升高。
(3)肌肉活檢顯示肌糖原累積、肌磷酸化酶缺乏。
6.Ⅵ型診斷依據
(1)症狀和體徵:肝大,可有低血糖發生。
(2)空腹或餐後注射胰高糖素不能使血糖升高。
(3)肝活檢糖原含量高,磷酸化酶活性低。白細胞中此酶活性低。
7.Ⅶ型診斷依據
(1)症狀和體徵:同Ⅴ型。
(2)肌肉活檢缺乏磷酸果糖激酶,紅細胞中此酶活性低。
8.磷酸酶b激酶缺乏診斷依據
①症狀和體徵:如肝大等。②測定白細胞或肝細胞酶活性降低。
9.Ⅹ型診斷依據
①肝大。②胰高糖素試驗陽性。③肝臟或肌肉活檢。
10.O型診斷依據
①症狀和體徵 ②胰高糖素試驗:空腹試驗無反應,餐後呈高血糖反應。③餐後肝臟活檢肝糖原含量低於肝濕重0.5%。④紅細胞糖原合成酶活性檢測。
鑑別診斷:
糖原貯積病主要應與其他的代謝障礙性疾病相鑑別, 鑑別的關鍵在於受累組織或器官的活檢、酶學檢查以及染色體檢查等。
實驗室檢查:
1.空腹血糖測定。
2.血總膽固醇、三醯甘油測定。
3.血乳酸測定、尿酸測定。
4.胰高糖素試驗。
5.肝功能轉氨酶測定。
其它輔助檢查:
依據病情應選做骨骼X線檢查、腹部B超、心電圖 超聲心動圖等。必要時做組織或器官病理活檢。
治療及預後
1.Ⅰ型
(1)防治低血糖:急性發作時立即靜脈注射25%葡萄糖,維持血糖於2.22~6.66mmol/L。每2~3小時進食高蛋白、低脂肪飲食1次。
(2)防治酸中毒:血乳酸高,應服碳酸氫鈉。
(3)防治感染。
(4)別嘌醇(別嘌呤醇)治療高尿酸血症。
2.Ⅱ型尚無有效療法。
3.Ⅲ型進食宜少量多餐,高蛋白飲食, 限制脂肪和總熱量 ,試用苯妥英(苯妥英鈉)防治低血糖。
4.Ⅴ型 ①避免疲勞和劇烈運動。②運動前預備葡萄糖或果糖或給予異丙腎上腺素。
5.Ⅵ型 宜高蛋白飲食, 少量多餐。苯妥英(苯妥英鈉)防治低血糖。
預後:
Ⅳ型無特效療法,預後差,多死於繼發感染。Ⅵ型隨著年齡增長,肝臟可縮小,預後較好。糖原貯積病Ⅰ型,Ⅴ型,Ⅶ型經過適當治療預後較好。
