
概述
 染色體和DNA
染色體和DNA分類
 染色體
染色體指致病基因位於常染色體上,按顯性遺傳規律所發之病。所謂狹隘性,即無論致病基因為純合狀態(兩個等位基因都是致病因)或雜合基因(等位基因中一個是致病基因,另一個是正常基因)都能導致發病。機體從上代的生殖細胞中獲得帶有致病基因的常染色體時,就能發病。常染色體顯性遺傳病患者中大多數為顯性基因雜合狀態(雜合子)。若為純合狀態(純合子)則病情嚴重,常致死(流產、死胎或新生兒期死亡)。
特徵:①每代都有患者出現,在連續世代中呈垂直分布。②遺傳無性別差異,男女受累機會相同。③雙親中有一患者,則子代中發病幾率為50%,若雙親均為患者則子代中發病率為75%。④患者子代中正常者,則其不攜帶致病基因。⑤患者的雙親中必有患者,除非病性輕微未被發現或患者系新的基因突變所致。
病症:①軟骨發育不全,其主要特徵為四肢短小畸形,可能系遺傳性侏儒症中最常見的類型。因長骨骺端軟骨細胞形成障礙,影響骨的長度,但骨的寬度仍然增長,而導致四肢短小,侏儒體型。②蜘蛛腳樣指綜合徵,其特徵為肢體過長,眼病和心血管異常。因長骨過度生長而呈身材細長體型,上下段比例失常,四肢長,尤其指、趾細長;肋骨異常呈漏斗胸;肌肉發育差,皮下脂肪少,關節鬆弛;晶狀體異位或重度近視;主動脈瓣關閉不全或主動脈瘤。③多發性神經纖維瘤病,本病為神經外胚層的病變,皮膚上有黃棕色的色素斑為典型的特殊體徵,呈卵圓形或環狀不一,直徑為1~5cm;還常伴有皮膚神經纖維瘤,呈多發性,較小,質柔軟,稀疏分布,大的神經纖維瘤常在外周神經或神經根上,可導致脊柱畸形。。
④遺傳性舞蹈病,本病為大腦中基底核的病變,主要表現為進行性痴呆和不自主的舞蹈動作。起病隱匿,僅是正常的面部動作和手勢增加,以後呈現舞蹈樣的不隨意運動,舞蹈樣動作緩慢,兩次動作的間歇期較長。
2.常染色體隱性遺傳病(autosomalrecessivedisorder)
致病基因在常染色體上,基因性狀是隱性的,即只有純合子時才顯示病狀。此種遺傳病父母雙方均為致病基因攜帶者,故多見於近親婚配者的子女。子代有1/4的機率患病,子女患病機率均等。許多遺傳代謝異常的疾病,屬常染色體隱性遺傳病。按照“一個基因、一個酶”(onegeneoneenzyme)或“一個順反子、一個多肽”(onecistrononepolypeptide)的概念,這些遺傳代謝病的酶或蛋白分子的異常,來自各自編碼基因的異常。常見的常染色體隱性遺傳病有溶酶體貯積症,如糖原貯積症、脂質貯積症、粘多糖貯積症;合成酶的缺陷如血γ球蛋白缺乏症、白化病;苯丙酮尿症、肝豆狀核變性(Wilson病)及半乳糖血症等。
3.線粒體病
線粒體病是遺傳缺損引起線粒體代謝酶缺陷,使ATP合成障礙、能量來源不足導致的一組異質性病變。Luft等(1962)首先報導一例線粒體肌病,生化嚴重證實為氧化磷酸化脫偶聯引起。Anderson(1981)測定人類線粒體DNA(mtDNA)全長序列,Holt(1988)首次作線粒體病患者發現mtDNA缺失,證實mtDNA突變是人類疾病的重要病因,建立了有別於孟德爾遺傳的線粒體遺傳的新概念。根據線粒體病變部位不同可分為:①線粒體肌病:線粒體病變侵犯骨骼肌為主;②線粒體腦肌病:病變同時侵犯骨骼肌和中樞神經系統;③線粒體腦病:病變侵犯中樞神經系統為主。
遺傳方式
 遺傳規律圖
遺傳規律圖除上述幾種基本遺傳方式外,尚有2種特殊情況:
(1)從性遺傳:從性遺傳睡性連鎖遺傳的表現都與性別有密切關係,但它們是兩種截然不同的遺傳方式。性連鎖遺傳的基因位於性染色體上,而從性遺傳的基因位於常染色體上,致病基因性質有顯性和隱性之別。這種常染色體上的基因所控制的性狀,在表現型上受性別影響而男女性分布比例或表現程度上的差別,這種遺傳方式稱為從性遺傳(sex-influrencedinheritance)。
原發性血色病(primaryhematochromatosis)可作從性遺傳方式的實例。本病為一種遺傳性鐵代謝障礙,其特徵為含鐵血黃素在組織中大量沉積,造成多種器官損害,典型症狀是皮膚色素沉著、肝硬化、糖尿病三聯綜合徵,症狀發生較遲,由於鐵質蓄積達到15-30g方產生症狀,所以80%病例在40歲以後發病。本病致病基因在常染色體上,但男性多於女性10-20倍,而且女性發病較遲,這是因為女性通過月經、妊娠和哺乳,一生順可喪失鐵10-35g,故難以表現鐵質沉著症狀。
遺傳性早禿(hereditaryalopecia)為常染色體顯性遺傳病,男性顯著多於女性,女性僅表現為頭髮稀疏,極少全禿,雜合子(Bb)男性會出現早禿;相反,女性雜合子(Bb)不出現早禿,只有純合子(BB)才出現早禿,這也是從性遺傳的一例。
(2)限性遺傳;一種遺傳性狀或遺傳病的致病基因位於常染色體或性染色體上,其性質可以是顯性或隱性,但由於性別限制,只在一種性別得以表現,而在另一性別完全不能表現,但這些基因都可以向後代傳遞,這種遺傳方式稱為限性遺傳(sex-limitedinheritance)。例如,子宮陰道積水(hydrometrocolpos)由常染色體隱性基因決定,因此,女性只有在純合子才表現相應症狀,男性雖有這種基因但不能表現該性狀,然而這些基因都向後代傳遞。
上述從性遺傳和限性遺傳特點可見,並非所有表現出性別差異的遺傳性狀或遺傳病都是性連鎖遺傳,在常染色體遺傳病中有時也可見到性別差異,應注意加以區別。
遺傳異質性
 遺傳規律表
遺傳規律表RP的遺傳方式具有遺傳異質性,即可以有AD、AR、XR連鎖遺傳,可能還有Y連鎖遺傳。遺傳方式不同的RP,一般其遺傳基礎也不同,因而伴隨的綜合徵的以及始發年齡、主要病情變化特徵(XR常伴高度近視,AR和AD多為低度近視)、病程進展(AD快,AR慢)、預後情況(AD較輕,AR致盲)也有差異,甚至還可區分為其他不同亞型。
現知,XL的RP2基因定位於Xp11.4-11.23,XL的PR3基因定位於Xp21.1,AD的RP基因定位於8p11-q21。因此,普遍認為RP是多個基因座位上RP基因所引起的一組具有臨床亞型的視網膜退行性病變的遺傳性疾病。
特殊的遺傳現象
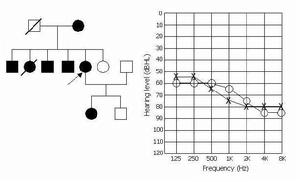 母系遺傳
母系遺傳由於mtDNA為母系遺傳,因此由mtDNA基因突變所致的Leber病也遵循母系遺傳的傳遞規律,即患者都與母親有關。男性患者的後代中尚未見有直接傳代者。但並非女性患者的後代全部發病,而且發病年齡也不一致;甚至一些女性患者本身表型正常,但可將本病傳給下一代。母系遺傳的特點:①母親將她的mtDNA傳遞給兒子和女兒,但只有女兒能將其mtDNA傳遞給下一代;②人的細胞里通常有上千個mtDNA拷貝,在突變體和正常mtDNA共存的細胞中,mtDNA在細胞的複製和分離過程中發生遺傳漂變,可導致子細胞出現三種基因型:純合的突變體mtDNA、純合的正常mtDNA、突變體和正常的mtDNA的雜合,這是由於mtDNA的遺傳不遵循孟德爾定律,被隨機分配到子細胞中所致;線粒體病發病有一閾值,只有當異常的mtDNA超過閾值時才發病。女性攜帶者的細胞內突變的mtDNA未達到閾值或在某種程度上受核影響而未發病,但仍可以通過mtDNA突變體向下代傳遞。女性患者細胞里mtDNA同樣可能存在雜合性,子女中得到較多突變mtDNA的個體發病,得到較少的病情較輕或不發病。
