
疾病簡介
 海洋性貧血
海洋性貧血 海洋性貧血又稱地中海貧血(Thalassemia),是一組遺傳性溶血性貧血。於1925年由Cooley和Lee首先描述,最早發現於地中海區域,當時稱為地中海貧血,國外稱海洋性貧血。實際上本病遍布世界各地,以地中海地區、中非洲、亞洲南太平洋地區發病較多。在中國以廣東、廣西、貴州、四川為多。
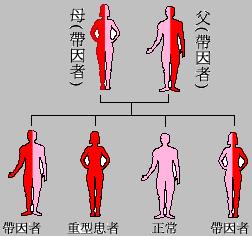
地中海貧血是一種嚴重的遺傳性疾病,其共同特點是由於珠蛋白基因的缺陷使血紅蛋白中的珠蛋白肽鏈有一種或幾種合成減少或不能合成。導致血紅蛋白的組成成分改變,本組疾病的臨床症狀輕重不一,大多表現為慢性進行性溶血性貧血。若夫妻為同型地中海型貧血的帶因者,每次懷孕,其子女有1/4的機會為正常,1/2的機會為帶因者,另1/4的機會為重型地中海型貧血患者。
病因病機
 海洋性貧血
海洋性貧血 本病是由於珠蛋白基因的缺失或點突變所致。組成珠蛋白的肽鏈有4種,即α、β、γ、δ鏈,分別由其相應的基因編碼,這些基因的缺失或點突變可造成各種肽鏈的合成障礙,致使血紅蛋白的組分改變。通常將地中海貧血分為α、β、δβ和δ等4種類型,其中以β和α地中海貧血較為常見。
β地中海貧血
 地中海貧血
地中海貧血 β 地貧基因突變較多迄今已發現的突變點達100多種國內已發現28種其中常見的突變有6種:
①β41-42(-TCTT)約占45%
②IVS-Ⅱ654(C→T),約占24%;
③β17(A→T);約占14%;
④TATA盒-28(A→T),約占9%;
⑤β71-72(+A),約占2%;
⑥β26(G→A),即HbE26,約占2%。
 地中海貧血
地中海貧血 輕型地貧是β0或β+地貧的雜合子狀態,β鏈的合成僅輕度減少,故其病理生理改變極輕微。中間型β地貧是一些β+地貧的雙重雜合子和某些地貧的變異型的純合子,或兩種不同變異型珠蛋白生成障礙性貧血的雙重雜合子狀態,其病理生理改變介於重型和輕型之間。
α地中海貧血
人類α珠蛋白基因簇位於16Pter-p13.3。每條染色體各有2個α珠蛋基因,一對染色體共有4個α珠蛋白基因。大多數α地中海貧血(簡稱α地貧)是由於α珠蛋白基因的缺失所致,少數由基因點突變造成。若僅是一條染色體上的一個α基因缺失或缺陷,則α鏈的合成部分受抑制,稱為α+地貧;若每一條染色體上的2個α基因均缺失或缺陷,稱為α0地貧。
1、重型α地貧
是α0地貧的純合子狀態,其4個α珠蛋白基因均缺失或缺陷,以致完全無α鏈生成,因而含有α鏈的HhA、HbA2和HbF的合成均減少。患者在胎兒期即發生大量γ鏈合成γ4(Hb Bart's)。Hb Bart's對氧的親合力極高,造成組織缺氧而引起胎兒水腫綜合徵。中間型和α地貧是α0和α+地貧的雜合子狀態,是由3個α珠蛋白基因缺失或缺陷所造成,患者僅能合成少量α鏈,其多餘的β鏈即合成HbH(β4)。HbH對氧親合力較高,又是一種不穩定血紅蛋白,容易在紅細胞內變性沉澱而形成包涵體,造成紅細胞膜僵硬而使紅細胞壽命縮短。
2、輕型α地貧
是α+地貧純合子或α0地貧雜合子狀態,它僅有2個α珠蛋白基因缺失或缺陷,故有相當數量的α鏈合成,病理生理改變輕微。靜止型α地貧是α+地貧雜合子狀態,它僅有一個α基因缺失或缺陷,α鏈的合成略為減少,病理生理改變非常輕微。
疾病類型
按照病因病機分類
1、α-地中海貧血
2、β-地中海貧血。
按照病情
1、重型出生數日即出現貧血、肝脾腫大進行性加重,黃疸,並有發育不良,其特殊表現有:頭大、眼距增寬、馬鞍鼻、前額突出、兩頰突出,其典型的表現是臀狀頭,長骨可骨折。骨骼改變是骨髓造血功能亢進、骨髓勝變寬、皮質變薄所致。少數患者在肋骨及脊椎之間發生胸腔腫塊,亦可見膽石症、下肢潰瘍。常見併發症有急性心包炎、繼發性脾功能亢進、繼發性血色病。
2、中間型輕度至中度貧血,患者大多可存活至成年。
3、輕型輕度貧血或無症狀,一般在調查家族史時發現。
臨床表現
β地中海貧血
 地中海貧血
地中海貧血 1、重型,又稱Cooley貧血。
患兒出生時無症狀,至3~12個月開始發病,呈慢性進行性貧血,面色蒼白,肝脾大,發育不良,常有輕度黃疽,症狀隨年齡增長而日益明顯。由於骨髓代償性增生導致骨骼變大、髓腔增寬,先發生於掌骨,以後為長骨和肋骨;1歲後顱骨改變明顯,表現為頭顱變大、額部隆起、顴高、鼻樑塌陷,兩眼距增寬,形成地中海貧血特殊面容。患兒常並發氣管炎或肺炎。當並發含鐵血黃素沉著症時,因過多的鐵沉著於心肌和其它臟器如肝、胰腺、腦垂體等而引起該臟器損害的相應症狀,其中最嚴重的是心力衰竭,它是貧血和鐵沉著造成心肌損害的結果,是導致患兒死亡的重要原因之一。本病如不治療,多於5歲前死亡。
實驗室檢查:外周血象呈小細胞低色素性貧血,紅細胞大小不等,中央淺染區擴大,出現異形、靶形、碎片紅細胞和有核紅細胞、點彩紅細胞、嗜多染性紅細胞、豪-周氏小體等;網織紅細胞正常或增高。骨髓象呈紅細胞系統增生明顯活躍,以中、晚幼紅細胞占多數,成熟紅細胞改變與外周血相同。紅細胞滲透脆性明顯減低。HbF含量明顯增高,大多>0.40,這是診斷重型β地貧的重要依據。顱骨X線片可見顱骨內外板變薄,板障增寬,在骨皮質間出現垂直短髮樣骨刺。
2、輕型
患者無症狀或輕度貧血,脾不大或輕度大。病程經過良好,能存活至老年。本病易被忽略,多在重型患者家族調查時被發現。
實驗室檢查:成熟紅細胞有輕度形態改變,紅細胞滲透脆勝正常或減低,血紅蛋白電泳顯示HbA2含量增高(0.035~0.060),這是本型的特點。HbF含量正常。
3、中間型
多於幼童期出現症狀,其臨床表現介於輕型和重型之間,中度貧血,脾臟輕或中度大,黃疽可有可無,骨骼改變較輕。
實驗室檢查:外周血象和骨髓象的改變如重型,紅細胞滲透脆性減低,HbF含量約為0.40~0.80,HbA2含量正常或增高。
α地中海貧血
1、靜止型患者無症狀。紅細胞形態正常,出生時臍帶血中Hb Bart's含量為0.01~0.02,但3個月後即消失。
2、輕型
患者無症狀。紅細胞形態有輕度改變,如大小不等、中央淺染、異形等;紅紐胞滲透脆性降低;變性珠蛋白小體陽性;HbA2和HbF含量正常或稍低。患兒臍血Hb Bart's含量為0.034~0.140,於生後6個月時完全消失。
3、中間型
又稱血紅蛋白H病。此型臨床表現差異較大,出現貧血的時間和貧血輕重不一。大多在嬰兒期以後逐漸出現貧血、疲乏無力、肝脾大、輕度黃疽;年齡較大患者可出現類似重型β地貧的特殊面容。合併呼吸道感染或服用氧化性藥物、抗瘧藥物等可誘發急性溶血而加重貧血,甚至發生溶血危象。
實驗室檢查:外周血象和骨髓象的改變類似重型β地貧;紅細胞滲透脆性減低;變性珠蛋白小體陽性;HbA2及HbF含量正常。出生時血液中含有約0.25Hb Bart's及少量HbH;隨年齡增長,HbH逐漸取代Hb Bart's,其含量約為0.024~0.44。包涵體生成試驗陽性。
4、重型
又稱Hb Bart's胎兒水腫綜合徵。胎兒常於30~40周時流產、死胎或娩出後半小時內死亡,胎兒呈重度貧血、黃疽、水腫、肝脾腫大、腹水、胸水。胎盤巨大且質脆。
實驗室檢查:外周血成熟紅細胞形態改變如重型β地貧,有核紅細胞和網織紅細胞明顯增高。血紅蛋白中幾乎全是HbBart's或同時有少量HbH,無HbA、HbA2和HbF。
併發症
 地中海貧血
地中海貧血 1、過量鐵質積聚:長期輸血會造成鐵質沉積而過量鐵質的積聚會對多個器官造成破壞。主要受影響的包括心臟、肝臟、胰臟和各個內分泌器官。病者會出現心臟衰竭、肝硬化、肝功能衰退、糖尿以及因為多種內分泌失調而變得身材矮小和發育不全等等。
2、輸血引起的反應:常見輸血時引起的不良反應包括發熱、發冷和出紅疹等。較嚴重的反應如急性溶血、氣管收縮和血壓下降等雖然甚少出現,但絕不能忽視。
3、經輸血而傳染的疾病:經輸血而傳染的主要是過濾性病毒疾病。雖然在輸血整個過程中,多重的預防措施已把傳染的機會減至極少,偶然亦有因輸血而感染丙型和B型肝炎的例子。愛滋病毒的傳染極為罕見。以上三種病毒中,只有B型肝炎可以靠有效的疫苗預防。
4、脾臟發大:在長期貧血和溶血的刺激下,不少重型和中型貧血病者都會出現脾臟變大的問題。過大脾臟會使貧血加劇和令病者需要接受更大量的輸血而導致更嚴重的鐵質積聚。及時把發大的脾臟切除往往能令情況改善。
5、膽石的形成:長期溶血令地中海貧血病人比一般人更容易患膽石。患有膽石的病人可能經常出現右上腹痛、皮膚、眼白變黃和茶色小便等的病徵。
6、除鐵藥的副作用:除鐵藥有時亦會影響視力、聽覺和骨骼生長。因此,除鐵藥物的注射份量應根據鐵質積聚的多少而定,切勿擅自把份量增加或減少。
診斷鑑別
根據臨床特點和實驗室檢查,結合陽性家族史,一般可作出診斷。有條件時、可作基因診斷。
骨髓象
診斷地中海貧血疾病需要注意增生活躍或明顯活躍、紅系細胞增生,主要以中幼、晚幼紅細胞增生為主,由於成熟紅細胞有靶形異形變,而骨髓細胞外鐵及鐵粒幼細胞增多。通過骨髓活檢顯示紅系增生顯著,幼紅細胞多聚集呈紅細胞造血島,在聚集團內有較多的原始紅細胞、早幼紅細胞,外周為中幼紅細胞、晚幼紅細胞,人們的骨髓間質無異常。
本病須與下列疾病鑑別。
1、缺鐵性貧血
輕型地中海貧血的臨床表現和紅細胞的形態改變與缺鐵性貧血有相似之處,故易被誤診。但缺鐵性貧血常有缺鐵誘因,血清鐵蛋白含量減低,骨髓外鐵粒幼紅細胞減少,紅細胞游離原葉琳升高,鐵劑治療有效等可資鑑別。
2、傳染性肝炎或肝硬化
因HbH病貧血較輕,還伴有肝脾腫大、黃疸,少數病例還可有肝功能損害,故易被誤診為黃疸型肝炎或肝硬化。但通過病史詢問、家族調查以及紅細胞形態觀察。
治療
 海洋性貧血
海洋性貧血 輕型地貧無需特殊治療。中間型和重型地貧應採取下列一種或數種方法給予治療。
1、一般治療注意休息和營養,積極預防感染。適當補充葉酸和維生素E。
2、輸血和去鐵治療此法是重要治療方法之一。
紅細胞少量輸注法僅適用於中間型α和β地貧,不主張用於重型β地貧。對於重型β地貧應從早期開始給予中、高量輸血,以使患兒生長發育接近正常和防止骨骼病變。
3、鐵鰲合劑常用去鐵胺(deferoxamine),可以增加鐵從尿液和糞便排出,但不能阻止胃腸道對鐵的吸收。
4、脾切除
脾切除對血紅查白H病和中間型β地貧的療效較好,對重型β地貧效果差。脾切除可致免疫功能減弱,應在5~6歲以後施行並嚴格掌握適應證。
5、造血幹細胞移植
異基因造血幹細胞移植是目前能根治重型β地貧的方法。如有HLA相配的造血幹細胞供者,;應作為治療重型β地貧的首選方法。
6、基因活化治療
套用化學藥物可增加γ基因表達或減少α基因表達,以改善β地貧的狀,已用於臨床的藥物有經基脲、5-氮雜胞苷(5~AZC)、阿糖胞苷、馬里蘭、異煙肼等,正在探索之中。
護理
1、地中海貧血患者往往體虛,要慎起居,適寒溫,注意預防外感;多進行戶外活動;呼吸新鮮空氣;進行適宜的體育鍛鍊,氣功鍛鍊,打太極拳等有助於增強體質和抗病能力。
2、飲食調理注意飲食調養。宜進食營養豐富的食物。
3、注意精神調養,對於虛勞血虛的預防有重要意義。七情過激,易使陰血暗耗,既是致病之因,又可加劇進程。所以本病患者應保持心情舒暢,樂觀。
預防
開展人群普查和遺傳諮詢、作好婚前指導以避免地貧基因攜帶者之間聯姻,對預防本病有重要意義。採用基因分析法進行產前診斷,可在妊娠早期對重型β和α地貧胎兒作出診斷並及時中止妊娠,以避免胎兒水腫綜合徵的發生和重型β地貧患者出生,是目前預防本病行之有效的方法。
對地中海貧血尚無有效治療方法,但地貧的發生完全可以通過婚前醫學檢查、孕前醫學檢查和產前醫學檢查這3個環節得到有效控制。對夫婦雙方為地貧基因攜帶者的孕婦,在妊娠早期(2~4個月)進行產前基因診斷,防止重型地貧兒出生,是最有效的預防措施。一般來說,如果夫婦雙方屬同一類型地貧患者,便有機會生下重型地貧患者,其兒女有1/4的機會完全正常,1/2的機會成為輕型地貧患者和1/4的機會成為重型地貧患者。如果本人及配偶屬α或β輕型貧血患者,則胎兒必須接受產前診斷,以證實胎兒是否屬重型地貧患者。
地中海貧血的預防方法
1、開展人群普查和遺傳諮詢:作好婚前指導以避免地貧基因攜帶者之間聯姻,對預防本病有重要意義。採用基因分析法進行產前診斷,可在妊娠早期對重型β和α地貧胎兒作出診斷並及時中止妊娠,以避免胎兒水腫綜合徵的發生和重型β地貧患者出生,是目前預防本病行之有效的方法。
2、補充維生素:人們要在日常生活中做好預防地中海貧血的工作,防止人們的健康和生活受到任何的威脅,地中海型貧血帶因者外表、成長與正常人無異。維他命之補充,同平常人,於醫師認為必要時投予。因外表不易查覺此病,常被給予鐵劑補血,應注意。
3、婚前醫學檢查:婚前醫學檢查包括對地中海貧血等遺傳病的檢查。地中海貧血的人群基因攜帶率達10%以上,遺傳風險很大,如果男女雙方都攜帶地中海貧血同類基因,婚後孕育後代有25%機率是正常兒,50%機率是地中海貧血基因攜帶者(即輕型地中海貧血患者),25%是重型地中海貧血患者。
飲食常識
地貧患者要適當補充葉酸和維生素,但是由於不少所謂多種維他命或“補血”藥都可能含有鐵質,地中海貧血病人亦不宜進食此類藥物以避免過量吸收鐵質。一些含鐵質量高食物如肝臟、牛扒、菠菜、蘋果等,應適量而不宜過量進食。
飲食要均衡營養。食物多樣,並以穀類為主。不要過分節制飲食,及時糾正偏食,要平衡膳食。葡萄酒可治療貧血。適量飲點葡萄酒,有助於恢復健康。因為酒中含有多種維生素,如維生B1、B2、C及B12等,都是人體所不可少的。
地中海貧血患者要少吃冷飲;不要吃剩菜剩飯,特別是隔夜飯菜更不能吃;夏季飲食要少食過於油膩的食品,為了保證足量蛋白質的攝入,可吃一些雞蛋、瘦肉、魚和豆腐;少購買涼拌蔬菜,如涼拌黃瓜、涼拌西紅柿、涼拌粉皮等,如想吃涼拌菜,一定要將蔬菜用開水反覆燙洗沖淨,佐料一定要乾淨新鮮,現用現做,切不可用已調製好的陳舊佐料。
病情較輕的地中海貧血患者可以在食物中增添一些含鐵的輔助食品即可加以糾正,而不必刻意服補血藥,其中含鐵豐富的食物有豬肝、瘦肉、蛋黃、綠葉蔬菜、土豆等都是地中海貧血患者可以多吃的食物。
地中海貧血患者不應該喝牛奶,一些正在服用補鐵藥物的患者也不宜喝牛奶,主要是因為各種食物中所含的鐵,必須在人體內的消化道中轉化成“亞鐵”才能被胃腸吸收和利用;而這一轉化極容易受牛奶中的高磷多鈣的影響,人體內原有的鐵能與牛奶中的鈣鹽、磷鹽相結合而變成不易溶化的含鐵化合物,從而不能被人體所利用,因此地中海貧血患者絕對不可以在康復期間喝牛奶以保證更好的康復。
患了地中海貧血的人日常生活中最好是不要喝茶,特別是在飯後。因為,喝茶會阻止人體對食物中鐵的吸收,不利於貧血症狀的改善。
貧血嚴重性
地中海貧血是一種遺傳性、溶血性貧血地中海貧血是一種嚴重的遺傳性貧血,是由於先天性的基因缺陷導致人體不能正常合成血紅蛋白,使紅血球在脾臟和骨髓里就很容易溶解、破碎,形成慢性、遺傳性的貧血。地中海貧血的最大危害來自於貧血。長期貧血會導致病人臟器功能減退,首先骨髓會過度造血,引起骨板變薄,導致病人骨質疏鬆,長期貧血會導致鐵的吸收過量,還會導致心功能衰竭、肝脾腫大等,這些因素累計起來就會影響到將來的生長發育。
地中海貧血是先天的,但是並不是一出生就有的。不管是重型的還是中間型的,剛出生時和正常孩子沒有什麼區別,但是重型的在3個月左右就慢慢出現貧血了,特別是高發區的小孩一旦出現了類似情況就應該想到可能是地中海貧血,中間型的可能在4-5歲的時候慢慢出現貧血,也要做相關檢查。需要強調的是雖然別的疾病也可能出現相關症狀,但是高發區應該尤其引起重視。
輕度地中海貧血患者就像正常人一樣,沒有什麼特別的不良反應,家人也無需太注意,跟平常生活一樣,不要進行特別的照顧。中、重度的地中海貧血會使患者出現貧血的症狀,渾身無力、營養不良、肝脾腫大、黃,隨著年齡增長會越來越嚴重。骨骼也會變大,導致頭顱變大、額部隆起、顴高、鼻樑塌陷、眼距寬的面容改變。還會出現支氣管炎和腎炎。各種臟器由於鐵質的沉積會有一定的損害,導致心力衰竭死亡。
