概述
 非特異性間質性肺炎
非特異性間質性肺炎在上述7種臨床病理類型中,以IPF最為常見,也是臨床醫師最為熟悉的,其臨床特徵和診斷標準也比較明確。但是,同樣也是臨床上比較常見的NSIP,因缺乏明確的臨床、影像、病理特徵,ATS/ERS共識報告中建議將其暫定為一種臨時性診斷,其疾病地位和轉歸有待進一步研究。作為病理學診斷的NSIP臨床上比較常見,可見於許多不同的疾病,包括已知原因的過敏性肺炎、風濕免疫疾病等。如果原因不明,則可稱為特發性NSIP。在IIP的分類中,確立NSIP這一
 非特異性間質性肺炎
非特異性間質性肺炎類型,其初衷是要把一組不能歸類於其他IIP類型的間質性肺炎識別出來。就像其名稱所提示的那樣,NSIP在臨床、影像和病理方面有許多“非特異性”特徵。最初有人擔心NSIP會成為一種“廢紙簍”診斷,凡是不能歸類於其他IIP的,都會扔到NSIP的範疇內。更為複雜的是,NSIP的病理改變也可見於未知原因的IIP。例如NSIP的病理改變與IPF的病理改變(普通型間質型肺炎,UIP)常常並存,有時難以鑑別。越來越多的資料表明,準確的鑑別診斷具有重要臨床意義,並不單純是一個學術上的或語義上的爭論,因為UIP/IPF的預後遠不如特發性NSIP。另外,特發性NSIP的富細胞型和纖維化型預後也不同,富細胞型的預後要好。隨著更多NSIP病例的發現,其疾病地位的確立,已是臨床上迫切需要解決的問題了。
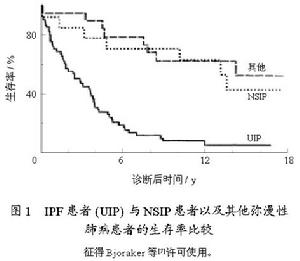
最近ATS的一個間質性肺病工作組發表了一份研究報告,通過大樣本病例分析,得出結論認為特發性NSIP是一種獨立存在的疾病(TravisWD,etal.Idiopathicnonspecificinterstitialpneumonia.ReportofanAmericanThoracicSocietyProject.AmJRespirCritCareMed,2008;177:1338-47)。
 非特異性間質性肺炎
非特異性間質性肺炎工作組邀請曾發表過特發性NSIP病例總結的作者參加研討會並提交各自的病例資料,然後統一由呼吸科、放射科和病理科醫師分別進行單獨評價和多學科評價,最後形成一致的臨床-影像-病理診斷(CRP診斷)。無論是在單獨評價還是在多學科共識診斷中,特發性NSIP診斷的確信程度被分為以下4個等級:肯定是(definite)、很可能是(probable)、可能不是(possible)和肯定不是(definitelynot)。“肯定是”或“肯定不是”表示強烈支持或否定NSIP診斷,“很可能是”表示支持NSIP診斷,“可能不是”表示不支持NSIP診斷。工作組對得出一致CRP診斷的NSIP病例進行分析後,總結出了特發性NSIP的臨床、影像和病理特徵。
結果在初步收到的305例病例中,112例因肺組織標本、HRCT、或臨床病史不充分、不完整、或相互矛盾而被排除。其餘193例的臨床、影像和病理資料符合入選標準,其中67例最終通過CRP診斷被認定為“肯定是”(17例)或“很可能是”(50例)NSIP。這67例最後診斷的確立全部符合以下3條標準:(1)外科肺活檢顯示NSIP病理類型(富細胞型或纖維化型);(2)HRCT表現與NSIP一致,不符合其他疾病例如UIP或慢性過敏性肺炎的表現;(3)在診斷時沒有其他慢性間質性肺病的臨床特徵,例如膠原血管病、藥物、或吸入抗原接觸史等。通過對67例病例進行分析,多學科研討會的專家達成共識認為,特發性NSIP是一個獨立的、具有特徵性臨床-影像-病理表現、與其他IIP不同的疾病實體。
特點
 非特異性間質性肺炎
非特異性間質性肺炎影像學特點:高分辨CT顯示雙肺對稱性毛玻璃影或雙肺肺泡腔的實變影。
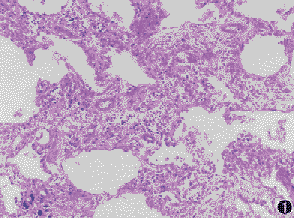
NSIP的組織學特徵包括不同程度的間質炎症和纖維化,具有一致性的外觀。富細胞型NSIP顯示輕度到中度的間質慢性炎症浸潤,基本沒有纖維化;纖維化型NSIP顯示間質增厚,為新舊一致的纖維化,肺泡結構完整,伴不同程度的細胞炎症。
發展過程
 相關藥物
相關藥物NSIP是最近提出的一個IPF的病理學類型。事實上這源於臨床實踐的需要。Katzentein等發現,有一部分病人在病理學上並不符合UIP、DIP或AIP的特徵,因而不能按照這三種類型來劃分,於是便另分為一組並稱之為“非特異性間質性肺炎”,含有不能分類的意思(nototherwisespecified)。然而越來越多的研究表明NSIP並非一個垃圾桶式的雜亂組合。這組病人有著相似的臨床和病理學表現,足以與其他類型一樣作為一個獨立的臨床病理學實體看待。
在有些文章中報導的“NISP”指的是常發生於感染了HIV病人的一種間質性肺炎[7],其臨床表現頗似卡氏肺囊蟲病,但找不到導致肺炎的病原體,而且病程為自限性,無需特異性治療。這與本文所指的NSIP不是一個概念。
病變特點
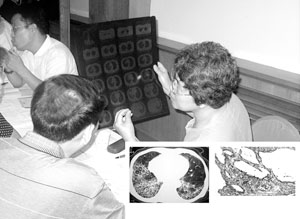 非特異性間質性肺炎的分析
非特異性間質性肺炎的分析NSIP在病理學上主要需與UIP相鑑別。NSIP的特點為:肺泡壁明顯增厚,含有不同程度的炎症與纖維化表現。病灶可呈片狀分布,但最重要的特徵是在病變時相上的一致性,即不同部位的病變似乎都是由發生在一個狹窄的時間段內的損傷引起的,並且共處於炎症纖維化進程中的某一階段,在同一標本上見不到象UIP那樣的新老病灶共存的現象。然而在不同病例之間,炎症與纖維化的程度和比例可能有很大差異,並可據此將病人分為3組。第1組只有細胞性炎症而幾乎沒有纖維化,第2組炎症與纖維化並存,第3組有緻密的間質膠原沉積,只有輕度炎症並缺乏活動性纖維化表現。3組約各占50%、40%和10%。總的來說NSIP對肺泡結構的破壞較輕,即使是第3組病人也極少見到蜂窩樣改變。有的病人可有BOOP樣改變,但根據定義,NSIP標本中的BOOP樣病灶應占總體病變的10%以下。
除了病理學上的特點以外,NSIP在臨床上亦有許多特徵與UIP相區別。在性別構成上,綜合幾組報導的121例男女比例為1∶1.2(55/66),但在最近發表的一組研究中,23例NSIP只有1例是男性。NSIP的發病年齡亦以中老年為主,多數病人在40歲以上,但也有20歲以下發病者。比較突出的一個特點為起病方式。NSIP相對呈亞急性,從出現症狀到診斷很少超過1年。Nagai等報導的31例NSIP甚至平均只花了2個月,我們的這3例入院時平均病程為4個多月,而實際上病人第一次就醫的時間比這還要短得多。相比之下,UIP起病較隱襲,就診時通常已有1~2年的病史。NSIP的臨床表現除了咳嗽、呼吸困難和雙下肺爆裂音以外,22%~33%的病人有發熱,但杵狀指很少見,為13%,這與UIP正好相反。Nagai等報導的64例UIP無一發熱,66%有杵狀指。本文的第2、3例病人有不同程度的低熱。部分NSIP的病例還伴有可能與病因相關的因素,如結締組織病、有機灰塵吸入以及過去急性肺損傷史。在合併結締組織病的病例,肺部表現可先於其他系統的症狀。本文第1例病人即伴有原發乾燥綜合徵,然而呼吸系統的症狀明顯重於口眼部症狀;第2例在發病前曾接觸有機粉塵(劣質羊毛)。這些因素的存在也提示NSIP在發病機制上可能與UIP不同。
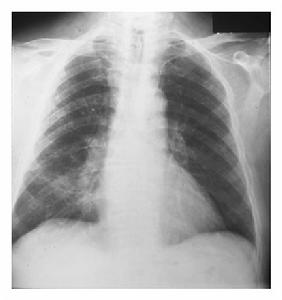 非特異性間質性肺炎底片
非特異性間質性肺炎底片對NSIP病人的BALF檢查發現細胞總數明顯增多,平均(4.4~4.5)×108/L;其淋巴細胞所占比例通常明顯高於UIP的病例,平均為37.7%~42.7%。本文3例的BALF淋巴細胞均升高,第2例淋巴細胞甚至占90%。最近Nagai等[4]報導,對NSIP患者BALF中T淋巴細胞亞群分析發現CD4/CD8比例明顯下降,在以炎症成分為主而纖維化較少的病例中,更可降至0.3;而在UIP這一比例平均值為1.65。我們對第2、3例進行了BALF的T淋巴細胞亞群分析,結果也是CD4/CD8明顯倒置,分別為0.25和0.227。這種T細胞亞群的變化頗令人感興趣,至於它對鑑別診斷的意義以及對判斷炎症程度與治療反應等方面的價值尚有待於進一步研究。
NSIP區別於UIP的最重要的臨床特點是預後明顯優於後者。NSIP對皮質激素的反應良好,絕大部分病人症狀能改善甚至完全緩解。本文第1例病人治療半年後,症狀、肺功能均明顯改善。目前報導NSIP的死亡率為6.5%~11%以下,而且死亡的都是伴有纖維化的病例。相反UIP對皮質激素多無反應而需要加免疫抑製劑,但也療效不佳,死亡率高達65%~75%,平均存活時間2.8~6年。長期以來的觀察發現,對治療有反應的IPF病例多為年輕患者、女性、HRCT上以磨玻璃樣變為主並且肺活檢為活動性炎症者。這些病例中可能有相當部分就是由NSIP構成的。
 學術研討會
學術研討會在過去,IPF是一個臨床概念,因此其中包含了多種病理學類型,也就包含了臨床表現上的異質性。對IPF組織形態學類型的劃分則在一定程度上揭示了其異質性的原因,並加深了人們對疾病本質的認識。由於NSIP、DIP/RBILD和AIP在臨床和病理學上都與UIP有明顯差異,也不符合人們對IPF的一般印象,越來越多的作者傾向於將IPF這一名稱保留給UIP的病例。NSIP由於其不同於UIP的臨床特點,尤其是預後明顯優於UIP,因此從病理學角度將NSIP與UIP明確區別有著重要的實踐指導意義。在臨床工作中,如遇到相對較年輕而且呈亞急性病程的IPF病人,伴有發熱或其他的全身性症狀,HRCT表現以片狀磨玻璃樣改變為主,BALF中淋巴細胞明顯增多且CD4/CD8比例倒置時,則可能是一例NSIP而不是UIP。這樣的病人可以通過胸肺活檢或經胸腔鏡肺活檢明確診斷。
臨床表現
 非特異性間質性肺炎
非特異性間質性肺炎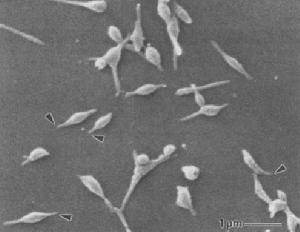 電子顯微鏡鏡下肺炎支原體
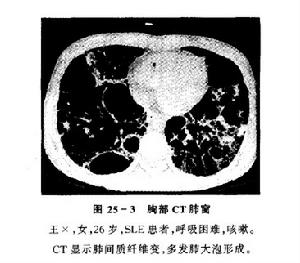
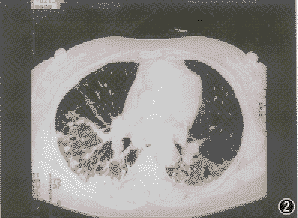
電子顯微鏡鏡下肺炎支原體三、咳嗽以夜間為重,偶爾有少量黃粘痰,入院前1個月出現活動後氣短,漸加重至上一層樓梯亦有症狀,同時伴胸背持續鈍痛。病程中有間斷髮熱,無關節肌肉痛史。體格檢查:輕度紫紺,無杵狀指,雙肺可聞爆裂音。血ANA、抗ds-DNA、RF及抗ENA均陰性。胸片示雙中下肺間質紋理厚,呈磨玻璃樣改變伴小斑片結節狀影。HRCT(圖4)見右肺中葉、左肺舌葉及雙肺下葉密度增高呈磨玻璃樣改變,雙肺支氣管血管紋理增厚,右肺中葉內側段支氣管擴張,雙側胸膜不規則增厚,縱隔內及雙肺門淋巴結多發鈣化。血氣分析PaO261.4mmHg。BALF細胞總數1.27×109/L,淋巴細胞占32%,中性細胞占47%,巨噬細胞占21%;T淋巴細胞亞群分析:CD417.6%,CD877.7%,CD4∶CD8=0.227。經支氣管肺活檢(TBLB)病理所見為少許血管及破碎的支氣管黏膜上皮呈急性及慢性炎。經電視引導下胸腔鏡肺活檢確診為非特異性間質性肺炎。
鑑別診斷
 非特異性間質性肺炎
非特異性間質性肺炎工作組專家認為,特發性NSIP與其他多種間質性肺病的臨床、影像和病理學界限常常是模糊的。研討會達成共識提出,NSIP在臨床、影像和病理學方面,最容易與以下疾病重疊:(1)過敏性肺炎;(2)隱原性機化性肺炎;(3)UIP/IPF;(4)呼吸性細支氣管炎相關間質性肺病。
越來越多的報導顯示,NSIP是各種結締組織病中間質性肺炎最常見的類型之一。在一些結締組織病患者,間質性肺病是最初的臨床表現,而且常常是NSIP。感染和藥物毒性在病理上可表現為NSIP。另外,在經CRP診斷為亞急性或慢性過敏性肺炎的病例中,有的在外科肺活檢上NSIP是唯一的或主要的形態學表現。這進一步說明了作為病理診斷的NSIP,其臨床疾病的異質性;因此強調在做出特發性NSIP之前,應充分進行鑑別診斷,排除繼發性NSIP。
 非特異性間質性肺炎
非特異性間質性肺炎1.臨床表現為呼吸困難和咳嗽,病程通常為6~7個月,女性患者多見,多為不吸菸者、多發生在50歲~60歲年齡段。大多數病例肺功能顯示限制性通氣障礙。
2.HRCT的主要特徵是:雙側、對稱、主要在下肺的網狀陰影,伴牽拉性支氣管擴張和下葉體積減少,通常為瀰漫性或胸膜下分布,但有時病變並不累及胸膜下肺組織。
3.NSIP的關鍵病理學特徵是間質病變的一致性,顯示從細胞型到纖維化型的病變過程。
4.特發性NSIP決大多數病例預後良好,5年死亡率預計低於18%。
治療方法
傳統方法治療間質性肺炎多採用激素類藥物來緩解患者的症狀,無法達到徹底根治間質性肺炎的目的,只能暫時控制病情,治標不治本容易反覆發作,長期服用易產生耐藥性,對肝、腎功能造成損害,毒副作用影響較大,危害了患者的身體健康。4S清肺平喘療法由軍事醫學科學研究院、國際科學研究院等多位專家歷經30多年聯合研發形成的一套全方位、多立體的綜合治療體系。療法包括清肺淨肺、PI活肺治療、植物神經綜合平衡和軍科細胞免疫技術四大步驟,其中軍科細胞免疫技術是多位專家在2008年諾貝爾醫學獎DC細胞技術基礎上獨創的治療手段,它直擊哮喘病的發病根源——炎症細胞,利用特定的靶向免疫激活技術抑制和清除哮喘發病根源,真真正正防止哮喘反覆發作。治療效果
1.當天症狀緩解。咳嗽、氣喘、呼吸困難、胸悶等症狀緩解。
2.3天內炎症消除、症狀消失,肺部和氣道功能恢復。
3.5——7天,免疫增強,炎症細胞消失,呼吸暢通,臨床治癒不復發。
研究方向
 非特異性間質性肺炎
非特異性間質性肺炎3.確定NSIP是否具有性別和種族傾向性;
4.明確特發性NSIP是否是一種自身免疫病;
5.與結締組織病相關的NSIP相比,特發性NSIP是否具有不同的表現、或不同的影像、病理特徵;
7.發現最佳治療藥物的臨床試驗。
相關詞條
呼吸內科疾病
| 研究範圍及進展呼吸內科是研究呼吸系統疾病的學科。它是既古老又年輕的學科,說它古老是因為,自從人類認識疾病以來,呼吸系統疾病就一直是危害人類健康的常見病和多發病,八十年代中期的統計資料表明,呼吸系統疾病仍然是導致死亡的主要疾病。在死亡的順序上排列第二。現在我們來認識一下這些可怕的疾病吧? |
