
概況?
 肺解剖圖
肺解剖圖?中文拼音:tè fā xìng fèi jiān zhì xiān wéi huà
英文參考:idiopathic pulmonary fibrosis,IPF
?特發性間質性肺炎(idiopathic interstitial pneumonia,IIP)又名特發性肺間質纖維化(idiopathic pulmonary fibrosis,IPF)是指原因不明的瀰漫性肺間質纖維化,為一種比較常見的肺疾病。可發生於任何年齡,患者以中、老年較多,多見於40~60歲之間。臨床特徵是出現進行性呼吸困難,X線顯示兩肺瀰漫性網狀結節狀陰影,肺功能檢查表明有限制性通氣功能障礙、彌散功能障礙和肺的順應性降低。病變特徵是,早期表現為脫屑性間質性肺炎,晚期呈現不同程度的間質纖維化和蜂窩肺。多數患者呈慢性經過,但本病預後不佳,病死率甚高,常因肺功能不全和心力衰竭而死亡。平均生存期為5~6年,也有存活10年以上者。少數急性型病例進展急劇,多在6個月內死亡。年齡愈小者,病程愈短。
疾病描述??
病因病機?
 呼吸系統解剖圖
呼吸系統解剖圖??本病確切的病因和發病機制不明,可能與自身免疫、遺傳因素和病毒感染有關。
目前較多的看法,認為特發性肺間質纖維化是一種自身免疫性疾病。其根據是本病常常與一些自身免疫性疾病,如類風濕性關節炎、硬皮病、系統性紅斑狼瘡等同時發生,而且在這些疾病中,肺部發生的病變與IPF的病變極其相似。IPF患者可出現高丙種球蛋白血症,升高的免疫球蛋白主要是IgG、IgM、IgA。部分病人可檢出自身抗體,尤其是抗核抗體,類風濕因子陽性率較高。患者支氣管肺泡灌洗液和血清中可檢出免疫複合物(IgG-抗原複合物及IgM-C3b-抗原複合物),循環中免疫複合物也可在肺泡毛細血管壁內沉積。免疫複合物激活肺巨噬細胞釋放趨化因子,引起肺組織內中性粒細胞、單核細胞和嗜酸性粒細胞浸潤,而且這些細胞能產生氧自由基,還能分泌一些蛋白酶,如膠原酶、彈性硬蛋白酶等引起肺組織損傷,巨噬細胞還可產生纖維連線蛋白(fibronectin),促進纖維細胞增生,形成纖維化。
本病亦有家族性,有些患者是雙胞胎,同家族的患者,蛋管異地居住多年,仍可發生同樣的疾病。遺傳連鎖的研究表明,家族性IPF發生的危險與免疫球蛋白的γ異型有關。此外,在IPF患者中,存在HLA-B8、HLA-B12、HLA-B15和HLA-DW3、HLA-DW6、HLA-DR2抗原者增多,均提示本病發生與遺傳因素有關。
約40%患者症狀發作時有流感樣表現及胸部症狀,也有證據表明患者發病前有接觸病毒史。有人報告,IPF患者血清中EB病毒抗體增加,可測出對病毒殼體抗原的IgA。提出EB病毒可能在IPF的病因學中起重要作用。
病理變化?
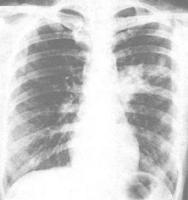 X線胸片
X線胸片??本病早期,病變表現為脫屑性間質性肺炎,Ⅱ型肺泡上皮增生,肺泡腔內充滿脫落的肺泡上皮細胞、肺泡巨噬細胞和淋巴細胞、中性粒細胞,有時可見透明膜形成。肺間質水腫,其中有灶性單核細胞、淋巴細胞和中性粒細胞浸潤,以及廣泛的纖維母細胞增生,肺泡間隔顯著增寬。隨著病情進展,肺泡腔和肺間質中的細胞逐漸減少。肺泡腔內炎性滲出物可發生機化,肺間質纖維組織逐漸增多,可發展至瀰漫性間質纖維化。細支氣管亦可見阻塞性細支氣管炎病變,肺小動脈可呈現閉塞性動脈炎。細支氣管和血管周圍的纖維化也極為明顯。到晚期,瀰漫性纖維化使肺體積縮小、質地變硬,細支氣管擴張呈小囊狀,肺泡也不規則擴張成小氣囊,小囊腔的囊壁破裂可形成較大的囊腔,這些囊被包裹於增生的纖維結締組織之中,形成蜂窩肺。
合併症?
?特發性肺間質纖維化常合併肺部感染而導致呼吸衰竭。患者也常合併肺氣腫、細支氣管擴張和肺源性心臟病。部分患者可出現自發性氣胸。少數病人可有肺性肥大性骨關節病。晚期特發性肺間質纖維化患者中,肺泡細胞癌、燕麥細胞癌、肺腺癌的發生率較高。
疾病分類?
 肺
肺?多年來,對IIP概念的理解一直存在差異,IIP的分類也經歷了一個不斷演化和修訂的過程。1969年Liebow等[7]首次提出了一組原因不明的瀰漫性間質性肺炎的概念,經典的病理組織學類型有5種,即:①尋常型(普通型)間質性肺炎(usual interstitial pneumonia,UIP);②脫屑性間質性肺炎(desquamative interstitial pneumonia,DIP);③閉塞性細支氣管炎性間質性肺炎(bronchiolitis obliterans with interstitial pneumonia,BOIP);④淋巴樣間質性肺炎(lymphoid interstitial pneumonia,LIP);⑤巨細胞間質性肺炎(giant cell interstitial pneumonia,GIP)。
1998年Katzenstein等[8]在以前研究的基礎上對這種瀰漫性的間質性肺疾病以特發性肺纖維化(idiopathic pulmonary fibrosis,IPF)命名,又重新提出了新的病理類型,包括:①普通型間質性肺炎(UIP);②脫屑性間質性肺炎(DIP)/呼吸性細支氣管炎並間質性肺疾病(RBILD);③急性間質性肺炎(acute interstitial pneumonia,AIP);④非特異性間質性肺炎(nonspecific interstitial pneumonia,NSIP)。
此分類將閉塞性細支氣管炎伴間質性肺炎(bronchiolitis obliterans with interstitial pneumonia,BIP)的概念被閉塞性細支氣管炎機化性肺炎(bronchiolitis obliterans organizing pneumonia,BOOP)所代替,而LIP和GIP這2個類型從IIP家族中被剔除,原因是發現LIP是一種與免疫缺陷相關的淋巴增殖性疾患,而GIP是重金屬塵肺的表現。除此之外,有的研究提出了呼吸性細支氣管炎伴間質性肺疾病類型(RBILD),有些國家把BOOP命名為隱原性機化性肺炎(COP)。
2000年ATS和ERS發表了有關IPF診斷和治療的多國專家的綜合意見[9],IPF的分類和診斷達成了新的國際共識,UIP是與IPF相一致的組織病理類型,因此IPF即特指UIP,而DIP、RBILD、NSIP和AIP等為不同的獨立疾病實體,它們與UIP/IPF一起同屬於特發性間質性肺炎(IIP)。
時隔2年,2002年ATS又發表了對ATS/ERS分類的修訂意見[10],對IIP的亞型重新界定,指出IIP除了包括UIP/IPF、NSIP、DIP、RBILD和AIP外,還應包括特發性LIP和隱原性機化性肺炎(cryptogenic organizing pneumonia,COP)。COP與特發性BOOP(IBOOP)為同一概念。新的ATS/ERS分類統一了既往病理和臨床對IIP概念和分類的不同看法和認識,有利於IIP的診治以及國際間的科研合作。當然,這個分類的適用性和合理性還有待實踐的檢驗和完善。具體分類:①UIP/IPF;②非特異性間質性肺炎(nonspecific interstitial pneumonia,NSIP);③隱原性機化性肺炎(cryptogenic organizing pneumonia,COP);④急性間質性肺炎(acute interstitial pneumonia,AIP);⑤呼吸性細支氣管炎性間質性肺疾病(respiratory bronchiolitisassociated interstitial lung disease,RBILD);⑥脫屑性間質性肺炎(desquamative interstitial pneumonia,DIP);⑦淋巴樣間質性肺炎(lymphoid interstitial pneumonia,LIP)。
ATS/ERS分類同時指出,IIP各型的診斷除臨床和影像學資料外,明確診斷依賴於VATS/開胸肺活檢,但最後的病理診斷應密切聯繫臨床資料和影像學,即臨床放射病理診斷(clinicradiologicpathologic diagnosis,CRP診斷)。單獨由臨床醫師、放射科醫師或病理科醫師作出診斷都有可能是片面的,應儘可能進行CRP診斷。長期以來,病理和臨床採用不同的分類標準和術語,使有關IIP各亞型的名詞概念相當混亂,不利於診治和研究。根據ATS/ERS分類,IIP的病理學分類與CRP診斷的對應關係見表1[11]。表1 特發性間質性肺炎的病理分類與臨床(略)
特發性肺纖維化(IPF)/隱原性間質纖維化性肺泡炎(CFA)非特異性間質性肺炎(NSIP) 非特異性間質性肺炎(NSIP)機化性肺炎(OP) 隱原性機化性肺炎(COP)瀰漫性肺泡損傷(DAD) 急性間質性肺炎(AIP)呼吸性細支氣管炎(RB)
呼吸性細支氣管炎伴間質性肺疾病(RBILD)脫屑性間質性肺炎(DIP) 脫屑性間質性肺炎(DIP)淋巴樣間質性肺炎(AIP) 淋巴樣間質性肺炎(AIP)
臨床特點?
普通型間質性肺炎
臨床特點:此型在IIP中最為常見(占65%左右),50歲以上的成年人多發,約2/3患者年齡大於60歲,男性多於女性。臨床表現為乾咳、呼吸困難等,多數患者可聞及吸氣性爆裂音,以雙肺底部最為明顯,三分之一以上的患者可見杵狀指。肺功能異常主要為中至重度限制性通氣功能障礙和(或)彌散功能障礙。實驗室檢查缺乏特徵性,10%~25%的患者血清抗核抗體(ANA)和類風濕因子(RF)陽性。
影像學特點:胸部X線片主要表現是在兩肺基底部和周邊部的網狀陰影,常為雙側、不對稱性,伴有肺容積減少。CT對UIP的診斷具有重要的意義,主要表現為兩肺片狀、以基底部為主的網狀陰影,可有少量毛玻璃狀影。在纖維化嚴重的區域,常有牽引性支氣管和細支氣管擴張,和(或)胸膜下的蜂窩樣改變。
病變特點
:肉眼觀察雙肺體積縮小,重量增加,質地較硬,髒層胸膜增厚,散在局灶性瘢痕,可見肺氣腫甚至肺大泡形成。切面呈雙肺瀰漫性實變區,輕重不一,嚴重受累處形成多房囊性結構,即蜂窩肺。低倍鏡下病變呈斑片狀分布,主要累及胸膜下及肺實質,間質炎症、纖維化和蜂窩肺改變輕重不一,新舊病變交雜分布,病變間可見正常肺組織。早期病變是肺泡間隔增寬充血,淋巴細胞、漿細胞和組織細胞與散在的中性粒細胞浸潤,伴有Ⅱ型肺泡上皮和細支氣管上皮增生,部分肺泡內可見巨噬細胞。纖維化區有數量不等的膠原纖維沉積,炎症細胞相對較少,肺泡間隔毛細血管床減少乃至完全消失,其間可形成假腺樣結構,內覆增生的Ⅱ型肺泡上皮。蜂窩肺改變的區域是由大小不等的囊性纖維氣腔所構成,被覆有細支氣管上皮細胞。在纖維化區和蜂窩肺區可見有呼吸性細支氣管、肺泡管以及重建的囊壁內有大量增生之平滑肌束,形成所謂“肌硬化”。除了上述提及的老病灶(膠原沉積的瘢痕灶)外,同時還有增生活躍的肌纖維母細胞和纖維母細胞,基質呈粘液樣,位於肺間質,突向被覆呼吸上皮的腔面,此結構稱為纖維母細胞灶(fibroblast foci)。總之,纖維母細胞灶、伴膠原沉積的瘢痕化,不同時相病變的共存和蜂窩肺病變是診斷UIP的重要依據,也是與IIP其他類型相區別的要點[2,8]。
非特異性間質性肺炎
臨床特點:本型的確切發病率尚不清楚,估計約為36/10萬人,首由Katzenstein等(1994)提出。爾後的研究表明NSIP有著相對特異的臨床和病理學表現,應該作為一個獨立的病理實體看待。2000年和2002年的ATS/ERS分類都認同了特發性NSIP(1NSIP)在IIP家族中的地位。NSIP發病以中老年為主,可發生於兒童,平均年齡49歲,起病隱匿或呈亞急性經過。其病因不清,部分患者可伴有某些潛在的結締組織疾病、有機粉塵的吸入、某些藥物反應以及急性肺損傷的緩解期等。臨床主要表現為漸進性呼吸困難和咳嗽。與UIP相比,大部分NSIP患者對皮質激素有較好的反應和相對較好的預後,5年內病死率為15%~20%。
影像學特點:高分辨CT顯示雙肺對稱性毛玻璃影或雙肺肺泡腔的實變影。
病變特點:主要病理學特徵為肺間質不同程度的炎症和纖維化。根據其間質炎細胞的數量和纖維化的程度,Katzenstein和Fiori將NS1P分成3型:①富於細胞型,約占50%,主要表現為間質的炎症,很少或幾乎無纖維化,其特點為肺泡間隔內淋巴細胞和漿細胞的混合浸潤,其炎性細胞浸潤的程度較UIP和DIP等其他類型的間質性肺病更為突出。與LIP相比,此型肺泡結構沒有明顯的破壞,漿細胞的浸潤數量更為突出。間質炎症常常伴有肺泡呼吸上皮的增生。②混合型,約占40%,間質有大量的慢性炎細胞浸潤和明顯的膠原纖維沉著。此型與UIP不易鑑別,區別的要點是本病全肺的病變相對一致,無蜂窩肺,部分可見纖維母細胞灶,但數量很少。③纖維化型,約占10%,肺間質以緻密的膠原纖維沉積為主,伴有輕微的炎症反應或者缺乏炎症。很少出現纖維母細胞灶,病變一致是不同於UIP的鑑別要點。
隱原性機化性肺炎
臨床特點:隱原性機化性肺炎是一種原因不明的機化性肺炎,Davison等(1983)首次提出。COP發病年齡以50~60歲為多,平均55歲,無性別差異,與吸菸無關。病程多在2~6個月以內,2/5的患者發病有類似流感的症狀,如咳嗽、發熱、周身不適、乏力和體重減輕等。常有吸氣末的爆裂音。常規實驗檢查無特異。肺功能主要表現為限制性通氣障礙,靜息和運動後的低氧血症是一個常見的特點。2/3的患者對皮質激素有較好的反應。
影像學特點:胸片表現為雙側瀰漫性肺泡影,肺容積正常,復發性和遊走性陰影常見,單側肺泡陰影罕見。高分辨CT顯示肺部斑片狀肺泡腔內實變、毛玻璃影、小結節陰影和支氣管壁的增厚和擴張,主要分布在肺周圍,尤其是肺下野。
病變特點:主要病理變化是呼吸性細支氣管及以下的小氣道和肺泡腔內有機化性肺炎改變,病變表現單一,時相一致,呈斑片狀和支氣管周圍分布。病變位於氣腔內,肺結構沒有破壞,增生的纖維母細胞/肌纖維母細胞灶通過肺泡間孔從一個肺泡到鄰近的肺泡形成蝴蝶樣的結構,蜂窩肺不常見。
急性間質性肺炎
臨床特點:急性間質性肺炎罕見,為肺的急性損傷性病變。起病急劇(數日至數周內),表現為發熱、咳嗽和氣急,繼之出現呼吸衰竭,酷似原因不明的特發性ARDS。平均發病年齡49歲,無明顯性別差異。常規實驗室檢查無特異性。AIP病死率極高(>60%),多數在1~2個月內死亡。
影像學特點:X線胸片顯示瀰漫、雙側性肺陰影,CT掃描表現為雙側對稱斑片狀毛玻璃影。這種改變與急性呼吸窘迫綜合徵(ARDS)類似。
病變特點:主要的病理改變為瀰漫性肺泡損傷的機化期改變。病變時相一致,肺泡間隔顯著增寬,增寬肺泡隔內有卵圓到梭形的纖維母細胞增生,散在淋巴細胞和漿細胞浸潤,肺泡Ⅱ型上皮增生,細支氣管上皮可有鱗狀化生。少數肺泡腔內有少量透明膜。這是與其他IIP鑑別的關鍵點。
呼吸性細支氣管炎性間質性肺疾病
臨床特點:本型罕見,其特點是呼吸性細支氣管炎伴發周圍的氣腔內大量含色素的巨噬細胞聚積,與DIP極為相似。發病年齡平均36歲,男性稍多於女性,迄今報導的病例均有吸菸史;臨床表現類似DIP,杵狀指(趾)少見,雙肺有爆裂音。與UIP相比,糖皮質類固醇治療有明顯的效果,預後較好。
影像學特點:高分辨CT掃描約2/3的患者顯示網狀結節影,缺乏毛玻璃樣改變。
病變特點:病理變化與DIP類似,不同點在於本病相對局限在呼吸性細支氣管及其周圍的氣腔,其內有大量含色素的巨噬細胞聚集,遠端氣腔不受累,並且有明顯的呼吸性細支氣管炎,肺泡間隔增厚和上皮化生等亦類似於DIP的表現。
脫屑性間質性肺炎
臨床特點:“脫屑”是指肺泡上皮脫落聚集在肺泡腔內的現象。本型肺泡腔內聚集的細胞不是肺泡上皮而是巨噬細胞,“脫屑”這個概念不準確,但現在一直延用此命名。DIP的治療和預後都較UIP為好,10年生存率大約為70%。DIP多見於有吸菸史者,平均發病年齡是42歲,男性多見,約為女性的2倍。大多數患者為亞急性起病(數周至數月)或隱匿,臨床表現與UIP類似,咳嗽和呼吸困難是最常見的症狀,半數患者有杵狀指。肺功能為限制性通氣障礙,伴有彌散功能降低和低氧血症。一般實驗室檢查無特殊發現。
影像學特點:20%的患者胸片接近正常。大約1/4的患者胸片和高分辨CT掃描顯示在中下肺野出現瀰漫的毛玻璃樣改變,後期也可出現線狀、網狀、結節狀間質影像。
病變特點:主要的組織學特點是彌慢性的肺泡內巨噬細胞聚集,均勻分布。這種變化在呼吸性細支氣管周圍尤為明顯,並彌散到遠端氣腔甚至整個肺實質。除了肺泡壁輕至中度增厚外,無纖維化瘢痕、蜂窩肺,纖維母細胞灶缺如或不明顯。間質有少量淋巴細胞和漿細胞浸潤。
特發性間質性肺炎蛋白質
目的套用蛋白質組學的方法篩選出特發性間質性肺炎(IIP)相關蛋白質,為闡明IIP的發病機制和預後提供理論依據。方法收集8例通過小切口開胸肺活檢獲取的新鮮肺組織及5例遠離肺部良性病變的肺組織,提取組織總蛋白,運用固相PH梯度(IPG)雙向凝膠電泳(2-DE)技術分離組織總蛋白,圖象分析軟體識別差異表達的蛋白質點,運用基質輔助電離解析飛行時間質譜(MALDI—TOF—MS)獲取肽質量指紋圖譜(PMF),檢索資料庫鑑定差異表達的蛋白質點,明確其生物學功能。結果建立雙向電泳圖譜,質譜分析鑑定出7個差異表達的蛋白質,分別為熱休克蛋白70、膜聯蛋白Ⅱ和觸珠蛋白等。結論建立了重複性較好的IIP與正常肺組織的雙向電泳圖譜,並鑑定出-些與IIP發病機制相關的蛋白質,為進-步尋找IIP特異分子標誌物和靶向治療打下堅實的基礎。
淋巴樣間質性肺炎
臨床特點:LIP為一組肺淋巴組織增生性疾病,原來報導的LIP,一部分是發生於肺的黏膜相關淋巴組織性淋巴瘤(MALT)。2002年的ATS/ERS分類重新將特發性LIP歸入IIP家族的原因,主要是考慮到在IIP的鑑別診斷中需要考慮LIP,而且其本身也是一種不明原因的間質性肺炎,且顯示與間質性肺病相似的臨床和影像學特點。在HIV感染的人群、其他免疫缺陷和自身免疫性疾病患者中相對常見,1/3患者伴有Sjo¨[]gren綜合徵。
影像學特點:X線胸片表現為實變和血管周圍浸潤影。
病變特點:肺間質中瀰漫性的淋巴細胞、漿細胞和組織細胞浸潤,具有生髮中心的淋巴濾泡常見。Ⅱ型肺泡上皮有增生,肺泡腔內巨噬細胞增多。肺泡內的機化和巨噬細胞的聚集少見或輕微。免疫球蛋白輕鏈染色顯示B細胞為多克隆性。鑑別診斷包括支氣管黏膜相關淋巴組織增生(瀰漫性淋巴組織增生)、結節樣淋巴組織增生、MALT性和小細胞淋巴瘤,以及NSIP、過敏性肺炎和UIP等。
預後及預防
本病預後不良,大部分患者因肺纖維化導致肺動脈高壓、肺源性心臟病和右心衰竭,存活時間僅3~5年。我國尚無確切的患病率,至今缺乏特效治療方法,嚴重威脅著人們的健康。近年,由於研究方法的改進,電視胸腔鏡(VATS)/開胸肺活檢的開展,在國外近年來本病已成為呼吸病理研究的熱點。國內相關報導也日益增多[1~6]。
結論?
IIP的診斷和分類對病理醫師是一個新的問題,病理醫師必須仔細閱片,密切聯繫臨床和影像學資料,才能做出正確的診斷。儘管IIP各型都表現為不同程度的間質炎症和纖維化,但每型都有各自的病變特點。在病變進程上,除UIP顯示病變進展不一致外(即新老病變交雜、病灶之間有接近正常的肺組織),其他各型都顯示病變在同一個階段。胸膜下的蜂窩肺主要見於UIP,其他各型不易見到或出現較晚。DIP主要表現為瀰漫性的肺泡內巨噬細胞聚集。RBILD的病理變化與DIP類似,不同點在於病變相對局限在呼吸性細支氣管及其周圍的氣腔,有明顯的呼吸性細支氣管炎。纖維母細胞灶主要見於UIP。AIP有透明膜形成,其他各型則無此變化。COP主要顯示呼吸性細支氣管及以下的小氣道和肺泡腔內有機化性肺炎改變,其他各型的BOOP樣改變較局限或缺乏。
參考文獻?
1. 王京嵐,陳毅德,孫慶華,等.急性間質性肺炎――附一例報告[J].中華內科雜誌,1997,36(1):744
2. 將昭實,劉鴻瑞.普通型間質性肺炎的病理診斷[J].中華結核和呼吸雜誌,2000,23(1):15
3. 留永健,朱元鈺,劉鴻瑞,等.非特異性間質性肺炎三例報告並文獻複習[J].中華結核和呼吸雜誌,2000,23(1):19
4. 易祥華,何國鈞,鄭迪,等.非特異性間質性肺炎八例臨床病理分析[J].中華結核和呼吸雜誌,2002,25(1):81
5. 黃蓉,劉鴻瑞,住源於.隱原性機化性肺炎一例[J].中華病理學雜誌,2004,33(2):181
6. 易祥華,李惠萍,何國鈞,等.普通型間質性肺炎的病理特徵及其與特發性非特異性間質性肺炎的鑑別診斷[J].中華病理學雜誌,2004,33(2):100
7. Liebow AA,Carrington CB.The interstitial pneumonias. In:Simon M,Potchen EJ,LeMay M,eds.Frontiers of pulmonary radiology[M].New York:Grune & Stratton,1969.102~141
8. Katzenstein AL,Myers JL.Idiopathic pulmonary fibrosis:clinical relevance of pathologic classification[J].Am J Respir Crit Care Med,1998,157:1301
9. American Thoracic Society/European Respiratory Society.American Thoracic Society/European Respiratory Society Idiopathic pulmonary fibrosis:diagnosis and treatment International Consensus statement[J].Am J Respir Crit Care Med,2000,161:646
10. American Thoracic Society/European Respiratory Society.American Thorcic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias[J].Am J Respir Crit Care Med,2002,165:277
11. 易祥華,劉鴻瑞.特發性間質性肺炎的分類和病理診斷[J].中華病理學雜誌,2004,33(2):171
12. Katzenstein AL,Fioril RF.Nonspecific interstitial pneumonia/fibrosis:histologic feature and clinical significance[J].AM J Surg Pathol,1994,18:136
13. Katzenstein AL,Myers JL.Nonspecific interstitial pneumonia and the other idiopathic interstitial pneumonia:classification and diagnostic criteria[J].AM J Surg Pathol,2000,24:1
14. Davison AG,Heard BE,McAllister WA,et al.Crytogenic organizing pneumonia[J].Q J Med,1983,207:382
15. Katzenstein AL,Myers JL.Idiopathic interstitial fibrosis:clinical relevance of pathologic classification[J].Am J Respir Crit,1998,157:1301
16. Myers JL,Veal CF Jr,Shin MS,et al.Respiratory bronchiolitis causing interstitial lung disease.A clinicopathologic study of six cases[J].Am Rev Respir Dis,1987,135:880
