疾病描述
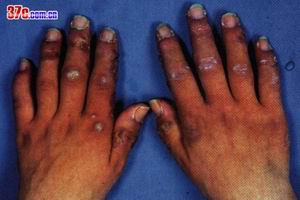 表現
表現症狀體徵
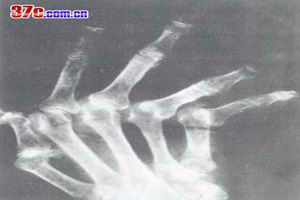 症狀
症狀病理生理
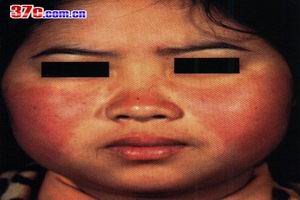 非特異性系統性壞死性小血管炎
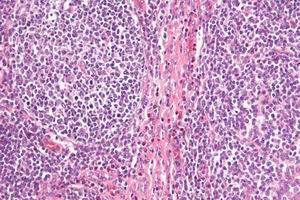
非特異性系統性壞死性小血管炎非特異性系統性壞死性小血管炎(nonspecificsystemicsmallvesselnecrotizingvasculitisSVN),既往亦稱顯微鏡下多發性微小動脈炎(microscopicpolyarteritis),現國際通用的病名為顯微鏡下多發性血管炎(microscopicpolyangiitisMPA),它是一種較常見的壞死性血管炎,主要病變為肺毛細血管炎,瀰漫性肺泡出血,肺泡間隔及間質有中性粒細胞浸潤,可見紅細胞及核塵,部分小血管有血栓形成及纖維素樣壞死。
診斷檢查
 診斷檢查
診斷檢查本病的肺部病變發生率為20%~30%,臨床有典型的三聯症:咯血、貧血和胸部X線片示肺泡出血徵象。咯血、貧血是常見症狀,甚至可發生致命性的大咯血,同時由於肺部氣體交換障礙,病人可發生嚴重的低氧血症,約12%病人因呼吸衰竭死亡。本病腎臟損害率高且症狀嚴重,腎臟受損的表現要多於肺部表現,約70%有腎功能損害,常合併腎功能衰竭而造成病人死亡,因腎功能衰竭造成的死亡率要高於呼吸衰竭。胸片表現為肺充血征,雙側肺野呈模糊陰影,屬肺泡炎性和充血性病變,間質有浸潤性改變,病變為雙側對稱性變化。
實驗室檢查:
有血沉、C-反應蛋白升高,血紅蛋白降低,支氣管肺泡灌洗液為血性液,巨噬細胞內有吞噬的含鐵血黃素。近年來ANCA研究發展對提高本病的診斷率有了極大的幫助,對本病有意義的ANCA抗體為核周ANCA(P-ANCA),其抗原為髓過氧化酶(MPO),其抗原抗體複合物為MPO-AN-CA,運用ELISA法和免疫螢光法結合可使MPA的診斷陽性率達到70%,尤其是高滴度的MPO-ANCA對確診MPA有非常高的價值。而且通過監測MPO-ANCA的滴度變化還可判斷疾病的輕重、觀察療效和指導治療,是目前本病診斷最新和最有前景的診斷技術。本病的確診仍需肺活檢,活檢組織的主要病變為肺毛細血管炎,瀰漫性肺泡出血,肺泡間隔及間質有中性粒細胞浸潤,可見紅細胞及核塵,部分小血管有血栓形成及纖維素樣壞死。
實驗室檢查:
1.實驗室檢查有血沉升高。C-反應蛋白升高。血紅蛋白降低。
2.支氣管肺泡灌洗液為血性液。巨噬細胞內有吞噬的含鐵血黃素。
3.抗中性粒細胞抗體近年來ANCA研究發展對提高本病的診斷率有了極大的幫助,對本病有意義的ANCA抗體為核周ANCA(P-ANCA),其抗原為髓過氧化酶(MPO),其抗原抗體複合物為MPO-AN-CA,運用ELISA法和免疫螢光法結合可使MPA的診斷陽性率達到70%,尤其是高滴度的MPO-ANCA對確診MPA有非常高的價值。
其他輔助檢查:
胸部X線片示肺泡出血徵象,雙側肺野呈模糊陰影,屬肺泡炎性和充血性病變,間質有浸潤性改變,病變為雙側對稱性變化。肺活檢組織的主要病變為肺毛細血管炎,瀰漫性肺泡出血,肺泡間隔及間質有中性粒細胞浸潤,可見紅細胞及核塵,部分小血管有血栓形成及纖維素樣壞死。
治療方案
 藥物治療
藥物治療(1)美國NIH1992年標準方案:
①初始治療:環磷醯胺2mg/(kg?d),最大5mg/(kg?d);潑尼松1mg/(kg?d)。
②維持治療:病人緩解後,環磷醯胺維持治療至少1年,每2~3個月降低劑量25mg。潑尼松連續套用4周后,在接下來的1~3個月內將劑量降至60mg以下,此後逐月減量,直至病人單獨套用環磷醯胺即可控制時停用。
③療效:完全緩解率75%,部分緩解率16%,兩年復發率50%。
④副作用:發生率43%,主要有套用環磷醯胺造成的出血性膀胱炎、膀胱癌以及激素引起的感染。
(2)英國Savage及其合作者方案:
①初始治療(4~6個月):環磷醯胺2mg/(kg?d),最大:150mg/d,年齡>60歲者:25mg/d,保持白細胞>4.0×109/L;潑尼松1mg/(kg?d),最大80mg/d,見效後至6個月時降至10mg/d。
②維持治療(6~24個月):硫唑嘌呤2mg/(kg?d);潑尼松5~10mg/d。
③升級治療:主要針對病情嚴重伴血Cr>500mmol/L和(或)肺出血病人。A.套用4.5%~5%人血白蛋白進行血漿置換治療,在14天內置換7~10次,總置換量60ml/kg。B.連用3天醋酸潑尼松龍:15mg/(kg?d),年齡<60歲病人可合用環磷醯胺25mg/(kg?d)。
④注意事項:大劑量環磷醯胺靜注療效不肯定;對不能耐受環磷醯胺者可用環孢素、甲氨蝶呤替換。
2.高劑量免疫球蛋白靜注(IVIG)治療IVIG療法是近年來治療血管炎性疾病的新方法,它來源於實驗室臨床實踐觀察,實驗觀察表明:IVIG可以以一種預測ANCA的遺傳性反應方式來中和ANCA誘發的反應;臨床研究發現:在其他自主免疫性疾病治療中套用IVIG方法取得了不同程度的緩解率,在一組套用免疫抑制劑和皮質激素無效的難治性病人中(包括11例MPA)套用IVIG治療2個月後的完全緩解率達50%,余者均部分緩解,治療前後病人的血沉、C-反應蛋白、ANCA、白細胞計數均明顯降低,1年後追蹤19例病人仍保持緩解,6例部分緩解,僅1例因毒血症死亡。同時上述病人伴用的免疫抑制劑和激素劑量也明顯降低(降低50%以上)。另外一組單獨套用IVIG治療的6例病人中(含3例MPA),1年後4例病人仍保持穩定。套用IVIG治療的副作用輕微,僅有少許皮疹、頭痛、關節痛,個別病人有一過性Cr增高,停藥後自行緩解。IVIG的套用劑量要大,目前尚無統一的標準,一般不低於2.0g/kg。
3.其他治療目前尚處於試驗階段的治療方法有:套用抗胸腺細胞球蛋白和抗T淋巴細胞單克隆抗體治療,具體劑量及用法尚處於探討階段而無確論,對於病毒(如B肝病毒)引起的血管炎可試用干擾素治療。目前常用的有Alphy及Garma型,用法:100萬U/d肌注或300萬U隔2天肌注,療程1~3個月。
預後及預防
 預防措施
預防措施病人可發生嚴重的低氧血症,約12%病人因呼吸衰竭死亡。本病腎臟損害率高且症狀嚴重,腎臟受損的表現要多於肺部表現,約70%有腎功能損害,常合併腎功能衰竭而造成病人死亡,因腎功能衰竭造成的死亡率要高於呼吸衰竭。
預防:
1.年齡>60歲及腎功能中度以上受損者,環磷醯胺劑量降至25mg/d。
2.如血白細胞總數<4.0×109/L或中性粒細胞<2.0×109/L,則立即停用免疫抑制劑。
3.皮質激素開始用量要大,維持使用時可換用常效製劑或日間高低劑量交替使用。
流行病學
 流行病學
流行病學呼吸內科疾病
| 研究範圍及進展呼吸內科是研究呼吸系統疾病的學科。它是既古老又年輕的學科,說它古老是因為,自從人類認識疾病以來,呼吸系統疾病就一直是危害人類健康的常見病和多發病,八十年代中期的統計資料表明,呼吸系統疾病仍然是導致死亡的主要疾病。在死亡的順序上排列第二。現在我們來認識一下這些可怕的疾病吧? |
