概述
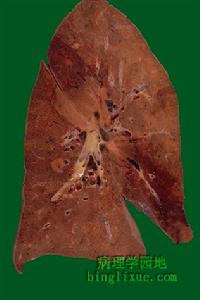 真菌性肉芽腫肺嗜酸細胞肉芽腫是指Langerhans組織細胞的增生性病變,以前包括在組織細胞增生症X(HX)之中。目前認為Langerhans細胞組織細胞增生症(Langerhans’cellhistiocvtosis,LCH)更為合宜,因為最近研究表明這組疾病的主要病變是Langerhans組織細胞的克隆性增生。表現為嬰幼兒的播散性急性病變(Letter-Siwe病)、慢性多灶性病變(Hand-Schuller-Christian)以及進展緩慢的局灶性病變(嗜酸細胞肉芽腫)。Letter-Siwe病常發生於2歲以內兒童,起病急、病情常較重,以廣泛的網狀內皮系統、骨骼和肺浸潤為特徵。Hand-Schüller-Christian病是一種比較慢性經過的疾病,主要發生在兒童和青年人,以肺和骨受累最多見。臨床上可出現經典性尿崩症,突眼以及溶骨性顱骨損害三聯征。這些病變可有相互重疊,有些兒童表現為孤立性肺部表現,某些成人表現為更具惡性傾向、播散性病變。肺LCH可以是多系統病變的一部分或只局限於肺臟(嗜酸細胞肉芽腫,原發性肺LCH)。原發性肺LCH是一種少見的與吸菸有關的間質性肺疾病,主要發生在年輕成人。少見的也有隻表現為孤立性溶骨病變;更為罕見的是多灶性或廣泛播散性病變,與兒科Letter-Siwe病相近。進展期病變相似於IPF;然而本病一般呈良性和遷延性臨床病程。儘管LCH與其他瀰漫性間質性肺疾病有某些相似之處,但作為一個獨立疾病,它有其不同於其他疾病的臨床、放射線學和病理學表現。
真菌性肉芽腫肺嗜酸細胞肉芽腫是指Langerhans組織細胞的增生性病變,以前包括在組織細胞增生症X(HX)之中。目前認為Langerhans細胞組織細胞增生症(Langerhans’cellhistiocvtosis,LCH)更為合宜,因為最近研究表明這組疾病的主要病變是Langerhans組織細胞的克隆性增生。表現為嬰幼兒的播散性急性病變(Letter-Siwe病)、慢性多灶性病變(Hand-Schuller-Christian)以及進展緩慢的局灶性病變(嗜酸細胞肉芽腫)。Letter-Siwe病常發生於2歲以內兒童,起病急、病情常較重,以廣泛的網狀內皮系統、骨骼和肺浸潤為特徵。Hand-Schüller-Christian病是一種比較慢性經過的疾病,主要發生在兒童和青年人,以肺和骨受累最多見。臨床上可出現經典性尿崩症,突眼以及溶骨性顱骨損害三聯征。這些病變可有相互重疊,有些兒童表現為孤立性肺部表現,某些成人表現為更具惡性傾向、播散性病變。肺LCH可以是多系統病變的一部分或只局限於肺臟(嗜酸細胞肉芽腫,原發性肺LCH)。原發性肺LCH是一種少見的與吸菸有關的間質性肺疾病,主要發生在年輕成人。少見的也有隻表現為孤立性溶骨病變;更為罕見的是多灶性或廣泛播散性病變,與兒科Letter-Siwe病相近。進展期病變相似於IPF;然而本病一般呈良性和遷延性臨床病程。儘管LCH與其他瀰漫性間質性肺疾病有某些相似之處,但作為一個獨立疾病,它有其不同於其他疾病的臨床、放射線學和病理學表現。體症
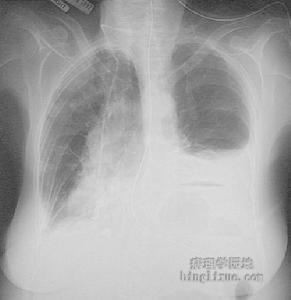 左胸膜腔積液LCH臨床表現多種多樣:有的因氣胸、或出現呼吸系統和全身症狀行胸部X線檢查而偶然發現。病人常表現為乾咳(50%~70%)、呼吸困難(40%)、胸疼(10%~21%)、疲勞(30%)、體重下降(20%~30%)以及發燒(15%)。50%病人有鼻炎病史。25%病人可出現反覆的胸膜痛及自發性氣胸。在未出現氣胸情況下罕見有胸膜肥厚和積液。偶有咯血報導(13%),此時應注意是否合併感染(如曲菌)或腫瘤。4%~20%病人可出現骨囊腫性損害,並產生局部疼痛或病理性骨折。由於未將全面骨檢查列為常規,所以發生骨損害病人的確切數量尚不清楚。骨受累症狀可在典型肺症狀之前出現,也可以是LCH的唯一表現。影像學表現無診斷意義。
左胸膜腔積液LCH臨床表現多種多樣:有的因氣胸、或出現呼吸系統和全身症狀行胸部X線檢查而偶然發現。病人常表現為乾咳(50%~70%)、呼吸困難(40%)、胸疼(10%~21%)、疲勞(30%)、體重下降(20%~30%)以及發燒(15%)。50%病人有鼻炎病史。25%病人可出現反覆的胸膜痛及自發性氣胸。在未出現氣胸情況下罕見有胸膜肥厚和積液。偶有咯血報導(13%),此時應注意是否合併感染(如曲菌)或腫瘤。4%~20%病人可出現骨囊腫性損害,並產生局部疼痛或病理性骨折。由於未將全面骨檢查列為常規,所以發生骨損害病人的確切數量尚不清楚。骨受累症狀可在典型肺症狀之前出現,也可以是LCH的唯一表現。影像學表現無診斷意義。
在多數情況下骨損害為單發,而且主要累及扁骨。中樞神經受累表現為尿崩症占15%,一般認為其提示預後不良。物理檢查常不明顯,爆裂音和杵狀指(趾)皆不常見。可發生繼發性肺動脈高壓;在進展期可見肺心病的表現。常規實驗室檢查常沒有意義,而且周圍血嗜酸細胞計數正常。生理
病因:不明的過敏反應性疾病,亦可能為病毒感染性疾病。
 肺纖維化生理:LCH發病機制不清,然而幾乎均有吸菸史這一事實提示吸菸可能為病因。本病發病機制的一個假說(鈴蟾肽假說)認為,鈴蟾肽樣肽產生增加發揮重要作用。鈴蟾肽是由神經內分泌細胞產生的一種神經肽,吸菸者肺臟中這種細胞增加。鈴蟾肽樣肽能趨化單核細胞、促進上皮和成纖維細胞有絲分裂,以及刺激細胞因子產生。因此這幾種顯著特徵支持有關這些肽類在LCH的炎症和纖維化中發揮作用這一假說。菸草糖蛋白和其他調節糖肽(如粒細胞-巨噬細胞集落刺激因子GM-CSF)可能在LCH發病機制中發揮潛在的重要作用。最近的有關研究集中在白細胞遊走的調節上。研究表明LCH發病機制包括調節白細胞和內皮細胞相互作用的黏附分子表達的改變。對中性粒細胞發揮重要作用、由內皮細胞表達的黏附分子為細胞間黏附分子-1(ICAM-1)。LCH病人的肺活檢標本中可見到Langerhans細胞表達ICAM-1。有趣的是其他白細胞黏附分子,如β1、β2整合素也有表達。這些改變的意義以及與LCH的相關性尚有待進一步闡述。另外,曾有研究提示病毒感染可作為全身性LCH的潛在病因。然而沒有可信服的資料提示病毒感染在LCH中發揮作用。在肺LCH也觀察到免疫功能異常,表現為BALF中IgG非特異增高、出現循環和組織親和性免疫複合物、以及T細胞功能異常,可能在本病的病理生理中有重要意義。然而這些改變也可能只代表了全身性免疫效應細胞激活。本病雖然不是單克隆疾病,但其常與淋巴瘤相伴提示其與惡性腫瘤的某種關係。目前,有理由認為肺HX可能是一種癌前病變。LCH早期炎症性病變以細支氣管為中心,含嗜酸細胞、淋巴細胞和中性粒細胞。其實LCH不屬於肉芽腫性疾病,而且病變又缺乏嗜酸細胞,所以曾用的舊名“嗜酸細胞肉芽腫”是不合適的。病變常累及肺小動脈和小靜脈,所以常描述為“沿支氣管血管分布"。LCH血管受累常見,但直到最近才得以定量評估。
肺纖維化生理:LCH發病機制不清,然而幾乎均有吸菸史這一事實提示吸菸可能為病因。本病發病機制的一個假說(鈴蟾肽假說)認為,鈴蟾肽樣肽產生增加發揮重要作用。鈴蟾肽是由神經內分泌細胞產生的一種神經肽,吸菸者肺臟中這種細胞增加。鈴蟾肽樣肽能趨化單核細胞、促進上皮和成纖維細胞有絲分裂,以及刺激細胞因子產生。因此這幾種顯著特徵支持有關這些肽類在LCH的炎症和纖維化中發揮作用這一假說。菸草糖蛋白和其他調節糖肽(如粒細胞-巨噬細胞集落刺激因子GM-CSF)可能在LCH發病機制中發揮潛在的重要作用。最近的有關研究集中在白細胞遊走的調節上。研究表明LCH發病機制包括調節白細胞和內皮細胞相互作用的黏附分子表達的改變。對中性粒細胞發揮重要作用、由內皮細胞表達的黏附分子為細胞間黏附分子-1(ICAM-1)。LCH病人的肺活檢標本中可見到Langerhans細胞表達ICAM-1。有趣的是其他白細胞黏附分子,如β1、β2整合素也有表達。這些改變的意義以及與LCH的相關性尚有待進一步闡述。另外,曾有研究提示病毒感染可作為全身性LCH的潛在病因。然而沒有可信服的資料提示病毒感染在LCH中發揮作用。在肺LCH也觀察到免疫功能異常,表現為BALF中IgG非特異增高、出現循環和組織親和性免疫複合物、以及T細胞功能異常,可能在本病的病理生理中有重要意義。然而這些改變也可能只代表了全身性免疫效應細胞激活。本病雖然不是單克隆疾病,但其常與淋巴瘤相伴提示其與惡性腫瘤的某種關係。目前,有理由認為肺HX可能是一種癌前病變。LCH早期炎症性病變以細支氣管為中心,含嗜酸細胞、淋巴細胞和中性粒細胞。其實LCH不屬於肉芽腫性疾病,而且病變又缺乏嗜酸細胞,所以曾用的舊名“嗜酸細胞肉芽腫”是不合適的。病變常累及肺小動脈和小靜脈,所以常描述為“沿支氣管血管分布"。LCH血管受累常見,但直到最近才得以定量評估。 正常肺顯微鏡顯示肺泡Travis注意到80%的活檢標本上有血管受累,還常見假脫屑性間質性肺炎(肺實質內在Langerhans細胞間充滿肺泡巨噬細胞)和呼吸性(吸菸者)細支氣管炎(細支氣管腔及周圍氣腔充滿含色素的巨噬細胞);除此之外,發現腔內纖維化常見(80%),以壁性整合、肺泡閉塞和腔內生芽為特點。其中59%為輕度,20%為中度,僅9%為重度。這些所見支持關於腔內纖維化是肺泡塌陷機制、並進展至肺纖維化以及肺重建的假說。間質纖維化和小囊腫形成以中、上野占優勢,發生在病變進展期;而且中、上野分布與IPF不同,後者病變多見於下野。病變進一步發展可廣泛累及支氣管血管周圍的肺實質並產生所謂“星狀病變”,為本病的特徵性改變。較陳舊病變細胞成分相對少,產生瀰漫性間質性病理改變,很難與其他終末期肺纖維化區別。囊腫形成的機制尚不清楚,可能是由於陳舊的星狀病變中心壞死所致;也可能是由於進展期支氣管血管病變遠端相對無血管區域的繼發性炎症性病灶而致;最後這些囊腫形成在一定程度上與由星狀病變而導致的近端氣道阻塞有關。LCH病理細胞類型是Langerhans細胞,由單核-巨噬細胞系分化而來。Langerhans細胞正常情況下可見於皮膚、網狀內皮系統、肺臟和胸膜,其胞漿染色淡、核仁大。電子顯微鏡可見典型的五層體胞漿包涵體或Birbeck顆粒(X小體)。雖然這種細胞也可見於健康吸菸者及其他肺病變(如IPF)或正常肺臟,但它的確是LCH的特點,表現為Langerhans細胞成群出現而且數量上明顯超過其他肺病變。但尚未建立起來LCH診斷的定量規範。
正常肺顯微鏡顯示肺泡Travis注意到80%的活檢標本上有血管受累,還常見假脫屑性間質性肺炎(肺實質內在Langerhans細胞間充滿肺泡巨噬細胞)和呼吸性(吸菸者)細支氣管炎(細支氣管腔及周圍氣腔充滿含色素的巨噬細胞);除此之外,發現腔內纖維化常見(80%),以壁性整合、肺泡閉塞和腔內生芽為特點。其中59%為輕度,20%為中度,僅9%為重度。這些所見支持關於腔內纖維化是肺泡塌陷機制、並進展至肺纖維化以及肺重建的假說。間質纖維化和小囊腫形成以中、上野占優勢,發生在病變進展期;而且中、上野分布與IPF不同,後者病變多見於下野。病變進一步發展可廣泛累及支氣管血管周圍的肺實質並產生所謂“星狀病變”,為本病的特徵性改變。較陳舊病變細胞成分相對少,產生瀰漫性間質性病理改變,很難與其他終末期肺纖維化區別。囊腫形成的機制尚不清楚,可能是由於陳舊的星狀病變中心壞死所致;也可能是由於進展期支氣管血管病變遠端相對無血管區域的繼發性炎症性病灶而致;最後這些囊腫形成在一定程度上與由星狀病變而導致的近端氣道阻塞有關。LCH病理細胞類型是Langerhans細胞,由單核-巨噬細胞系分化而來。Langerhans細胞正常情況下可見於皮膚、網狀內皮系統、肺臟和胸膜,其胞漿染色淡、核仁大。電子顯微鏡可見典型的五層體胞漿包涵體或Birbeck顆粒(X小體)。雖然這種細胞也可見於健康吸菸者及其他肺病變(如IPF)或正常肺臟,但它的確是LCH的特點,表現為Langerhans細胞成群出現而且數量上明顯超過其他肺病變。但尚未建立起來LCH診斷的定量規範。檢查
診斷:病史及物理檢查是對疑診肺LCH病人診斷評估的第一步,不幸的是其症狀和體徵常無特異性,而且常提示其他更常見的肺病變,如50歲的吸菸者出現氣喘、咳嗽和呼吸困難,COPD比LCH更多見;然而,當出現反覆氣胸、尿崩症及骨痛時則對診斷有幫助。吸菸史常見,但卻不是必需的病史表現,因為LCH確實可見於不吸菸病人。大多數病人LCH的診斷評估都在發現胸部影像學異常以後。例如CT表現典型則具有診斷意義,所以對疑診病人均應行CT檢查。對於所有疑診LCH的瀰漫性ILD,建議在活檢前行HRCT檢查。明顯的CT特徵以及合宜的臨床背景資料足可以免去組織學證實。然而值得注意是LCH胸部CT表現常不典型,所以,需與淋巴管平滑肌肌瘤病、結節性硬化症、過敏性肺泡炎、結節病以及IPF的影像混淆。在這種情況下應進一步進行其他診斷手段。BAL對疑診LCH有診斷價值。細胞總數增加(同吸菸者一樣);常見中性和嗜酸粒細胞略有增加;活動性病變淋巴細胞總數也可以增加,而且CD4/CD8比值下降;BALF中Langerhans細胞可由針對S-100蛋白或花生凝集素的特殊染色來認定。這些細胞呈OKT-6(CD-1)陽性,可用特異性單克隆抗體(MT-1)識別;在電鏡下含特徵性的Birbeck或五層體小體。目前尚沒有以BALF中Langerhans細胞數為依據的LCH確定性診斷標準。BALF細胞分數中Langerhans細胞也可見於其他疾病(現在吸菸者,其他ILD或細支氣管肺泡癌),甚至可見於正常人。所以僅只出現Langerhans細胞診斷價值不足。為求得組織學證據,TBLB足以確定診斷。取材錯誤和組織不足是造成假陰性或無診斷價值的主要原因。開胸或電視導引下經胸腔鏡肺活檢效果更確切而且手術危險性因素也降到了最低。在疑難病例可用單克隆抗體CD-1(OKT-6)行免疫染色以區別Langerhans細胞和其他組織細胞,對診斷有輔助作用。當出現廣泛纖維化的進行性病變時,組織標本和BALF中Langerhans細胞顯著減少,造成診斷困難。在多數情況下,結合TBLB、BALF分析,輔以組織和BALF中CD-1陽性細胞測定常足以得出正確診斷。
實驗室檢查:嗜酸性粒細胞數常規實驗室檢查沒有意義,而且周圍血嗜酸細胞計數正常。
其他輔助檢查:
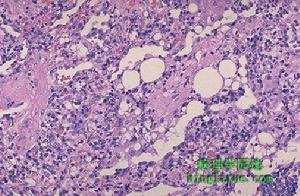 外源性類脂性肺炎1.胸部X線所見雖不具有診斷意義但很具有特徵性。如果出現邊界不清小結節影(2~10mm)、網狀結節影、上野囊狀變或蜂窩肺、肺容積不變以及肋膈角不受累等聯合表現則對本病診斷具有高度特異性。典型的網狀結節影見於中上野,與病理結果相一致。肺總量多正常,但也可見肺過度充氣和肺容積縮小。除LCH外,淋巴平滑肌瘤病、結節性硬化症、慢性過敏性肺(泡)炎、Ⅲ期結節病、縮窄性細支氣管炎以及任何有肺氣腫的ILD均可表現為肺容積增大。LCH的影像學標誌為小囊腫和小結節影,偶爾可見粟粒樣病變。LCH的肺門或縱隔淋巴結腫大罕見,如有腫大第二診斷應考慮惡性腫瘤。本病的原發胸膜受累少見,胸膜肥厚常是由於治療氣胸所致。骨損害可以發生於任何骨骼,包括肋骨。極罕見的情況下,病人只表現為孤立的肺部結節而在活檢中證實為LCH。
外源性類脂性肺炎1.胸部X線所見雖不具有診斷意義但很具有特徵性。如果出現邊界不清小結節影(2~10mm)、網狀結節影、上野囊狀變或蜂窩肺、肺容積不變以及肋膈角不受累等聯合表現則對本病診斷具有高度特異性。典型的網狀結節影見於中上野,與病理結果相一致。肺總量多正常,但也可見肺過度充氣和肺容積縮小。除LCH外,淋巴平滑肌瘤病、結節性硬化症、慢性過敏性肺(泡)炎、Ⅲ期結節病、縮窄性細支氣管炎以及任何有肺氣腫的ILD均可表現為肺容積增大。LCH的影像學標誌為小囊腫和小結節影,偶爾可見粟粒樣病變。LCH的肺門或縱隔淋巴結腫大罕見,如有腫大第二診斷應考慮惡性腫瘤。本病的原發胸膜受累少見,胸膜肥厚常是由於治療氣胸所致。骨損害可以發生於任何骨骼,包括肋骨。極罕見的情況下,病人只表現為孤立的肺部結節而在活檢中證實為LCH。
2.胸部的CT檢查當一年輕吸菸者,同時出現中上野分布為主的多發囊腫和小結節影時非常具有特徵性,可以考慮診斷LCH。小結節界限可以清晰或不清,偶爾可以較大而且形態奇異,進展期可見蜂窩肺改變。系列CT掃描可以觀察到,在一段時間內結節影經歷空洞變並向囊性變進展的過程。形成囊腫的程度在常規X線上常被低估,可以解釋在薄層CT常規套用以前文獻報告的很多所謂“自然緩解”病例。
3.磁共振(MRI)檢查MRI在LCH套用僅限於評估骨骼和CNS病變。
4.肺功能檢查LCH病人可表現出各種形式的肺功能改變,包括正常、阻塞性、限制性或混合性。一般來說,肺總量不變而且氣流幾乎正常;常見彌散功能不成比例地減低,提示有肺血管受累;少數病人出現氣流受限,而且有時與氣道反應性增高有關,經支氣管擴張劑治療後能得到明顯改善。當出現氣道反應性增高時則可能反映了有COPD共存。LCH出現典型的哮喘表現者不常見。Craussman等回顧的23例LCH發現,肺功能改變存在二個主要亞組。第一組表現為肺總量正常,氣流正常或近乎正常。此組病人行肺機械學測定時彈性回縮力正常。第二組表現為以限制性病變為主,肺總量減少和彈性回縮力增加。二者彌散功能均明顯減低,限制性病變組常表現為較長病程。二個亞組靜息狀態下平均AaDO2梯度均為正常,然而有5例嚴重病變病人AaDO2顯著增高且需要給氧治療。靜息狀態下pH和PaCO2多正常,所以靜息狀態下動脈血氣對病變評估很不敏感。
5.運動試驗臨床上LCH病人通常表現為活動受限及運動耐力下降,且與肺功能異常不成比例。在橫斷面上對23例LCH研究中發現,以作功或以運動極量氧耗量(VO2)評估的運動能力明顯降低,分別為預計值的54±4%和44±3%;極量運動時每搏氧輸出量降低到56±3%,無氧域降低到VO2max預計值的33±1%。最大通氣應答(VEmax,83±5%)超過了最大作功水平。最大通氣應答不受限,而且VE遠低於預計的通氣極限值。氣體交換異常表現在隨運動的增加而出現AaDO2增大。反映肺血管功能的指標VD/VT在多數病人出現異常增高或不下降,提示病變過程中或病理性或功能性累及到了肺血管。研究表明LCH病人運動耐力下降是由機械因素和肺血管受累共同引起的。鑑別診斷
本病首先與具有瀰漫性結節類型的肺結節病相鑑別,其次與特發性肺間質纖維化、慢性外源性過敏性肺泡炎和瀰漫性肺泡細胞癌相鑑別。
治療方案
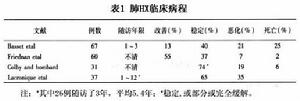 肺嗜酸細胞組織細胞增生症LCH自然病史差異很大,有些症狀自然緩解,有些則進展至終末期纖維性肺病(表1)。多數病人繼續吸菸時病情進展,而停止吸菸後病情緩解,所以應強調患者戒菸。皮質激素在LCH治療中未顯出任何治療價值;細胞毒製劑可能對治療播散性病變有一定價值,但對肺HX病人未見到益處。放射治療對骨病變可起緩解作用,對肺部表現無效。肺移植對於終末期病變可以考慮。將來,基因、單克隆抗體以及以細胞因子為基礎的療法可望成為潛在治療手段。因為本病有血管受累,而且偶爾出現肺動脈高壓,容易考慮到對有症狀病人行血管擴張劑治療,但這些療法仍是推測。
肺嗜酸細胞組織細胞增生症LCH自然病史差異很大,有些症狀自然緩解,有些則進展至終末期纖維性肺病(表1)。多數病人繼續吸菸時病情進展,而停止吸菸後病情緩解,所以應強調患者戒菸。皮質激素在LCH治療中未顯出任何治療價值;細胞毒製劑可能對治療播散性病變有一定價值,但對肺HX病人未見到益處。放射治療對骨病變可起緩解作用,對肺部表現無效。肺移植對於終末期病變可以考慮。將來,基因、單克隆抗體以及以細胞因子為基礎的療法可望成為潛在治療手段。因為本病有血管受累,而且偶爾出現肺動脈高壓,容易考慮到對有症狀病人行血管擴張劑治療,但這些療法仍是推測。併發症
常並發氣胸和合併感染(如曲菌)或腫瘤。
預後與預防
預後:此病多數預後良好,部分不治自愈,但累及多臟器者預後不佳。多數患者發展成瀰漫性肺間質纖維化,最後死於呼吸衰竭。
預防:由於吸菸與肺組織細胞增生症有著密切相關性,因此對於此類病人應盡最大努力勸阻病人戒菸。
流行病學
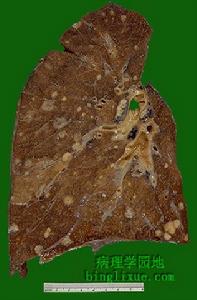 灰白色結節狀腫塊LCH確切發生率尚不清楚。很可能有些病例誤診為特發性肺纖維化(IPF)。LCH沒有職業和地理分布傾向。在一組研究中發現農民、伐木工人和接觸家養動物者發病偏多,分別為21%、25%和77%。值得注意的是幾乎所有報告病例均有吸菸史。因此認為吸菸可能是一病因因素。其他與吸菸相關的瀰漫性肺實質疾病,還有呼吸性細支氣管炎,間質性肺疾病/脫屑性間質性肺炎(RBILD/DIP)。LCH可發生於任何年齡組,但大多見於年輕人(20~40歲)。早期文獻提示男性偏多,然而近期文獻表明男女發病相同,而且中年發病有所增高;如果確有改變的話,這則反映了當今社會婦女吸菸人數增多。種族因素在發病機制中有重要作用,白種人比非洲和亞洲後裔發病多,後者本病罕見。有報導LCH與很多惡性腫瘤相關,而且可能是一種癌前病變。Sadoun和同事報導了95例HX中有5例發生支氣管肺癌。Tomashefsi和同事發現21例病人中有10例發展成為惡性(n=9)或良性腫瘤,其中3例肺癌、5例肺外腫瘤、2例淋巴瘤、1例肺類癌及1例縱隔神經節神經瘤;2例出現兩種不同惡性腫瘤。2例先於、3例後於、3例同時於LCH診斷。Hodgkin病或非Hodgkin淋巴瘤均有與LCH相伴發病的報導。出現這些腫瘤,吸菸可能是一種致病因素,所以吸菸在LCH發病中的作用難以確定。
灰白色結節狀腫塊LCH確切發生率尚不清楚。很可能有些病例誤診為特發性肺纖維化(IPF)。LCH沒有職業和地理分布傾向。在一組研究中發現農民、伐木工人和接觸家養動物者發病偏多,分別為21%、25%和77%。值得注意的是幾乎所有報告病例均有吸菸史。因此認為吸菸可能是一病因因素。其他與吸菸相關的瀰漫性肺實質疾病,還有呼吸性細支氣管炎,間質性肺疾病/脫屑性間質性肺炎(RBILD/DIP)。LCH可發生於任何年齡組,但大多見於年輕人(20~40歲)。早期文獻提示男性偏多,然而近期文獻表明男女發病相同,而且中年發病有所增高;如果確有改變的話,這則反映了當今社會婦女吸菸人數增多。種族因素在發病機制中有重要作用,白種人比非洲和亞洲後裔發病多,後者本病罕見。有報導LCH與很多惡性腫瘤相關,而且可能是一種癌前病變。Sadoun和同事報導了95例HX中有5例發生支氣管肺癌。Tomashefsi和同事發現21例病人中有10例發展成為惡性(n=9)或良性腫瘤,其中3例肺癌、5例肺外腫瘤、2例淋巴瘤、1例肺類癌及1例縱隔神經節神經瘤;2例出現兩種不同惡性腫瘤。2例先於、3例後於、3例同時於LCH診斷。Hodgkin病或非Hodgkin淋巴瘤均有與LCH相伴發病的報導。出現這些腫瘤,吸菸可能是一種致病因素,所以吸菸在LCH發病中的作用難以確定。相關疾病
特發性肺間質纖維化
慢性外源性過敏性肺泡炎
瀰漫性肺泡細胞癌
相關詞條
