概述
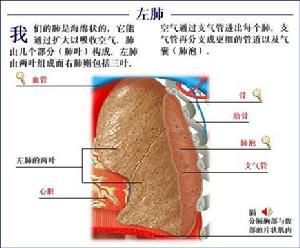 抗胰蛋白酶缺乏
抗胰蛋白酶缺乏自1963年以來laurell與Eriksson發現α1-抗胰蛋白酶(α1-antitypsin,簡稱α1-AT)缺乏與肺氣腫有密切關係,認為α1-AT缺乏是家族性肺氣腫的主要原因。發病年齡早,呼吸道症狀出現於30~40歲,早期症狀為活動後呼吸困難,多有咳嗽和反覆呼吸道感染,體檢見病人有過度消瘦,呼吸音低。α1-AT的缺乏與嬰幼兒肝硬化、肝癌的發病均有密切關係。
疾病體徵
 抗胰蛋白酶缺乏
抗胰蛋白酶缺乏病因
α1-AT的含量是由1組常染色體等位基因的不同類型所決定的,其基因座命名為Pi。在遺傳過程中,由兩個M型等位基因組成的純合子PiMM為正常的表現型,PiMM是最常見的正常功能基因型。PiZZ是α1-AT嚴重缺失的等位基因,純合子為ZZ,血清中α1-AT含量是正常人的15%~20%,這種人常發生阻塞性肺氣腫及幼年型肝硬化,PiS等位基因的純合子SS個體血清中α1-AT含量缺乏的占正常人的60%,這種人亦有患肺氣腫的傾向;此外其他表現型如MZ、SZ等雜合子也有α1-AT缺乏,其中有人發生肺氣腫和肝硬化。
發病機理
 抗胰蛋白酶缺乏
抗胰蛋白酶缺乏1.α1-AT的正常代謝α1-AT是由肝細胞分泌的糖蛋白,分子量為45000~56000萬(52Kda),α1-AT釋放入血漿後構成α1-AT球蛋白的主要成分,正常人(PiMM型)血漿中α1-AT濃度為1.3mg/ml,不同Pi型個體濃度有所差異。α1-AT是人類血漿中最主要的一種蛋白酶抑制劑(proteaseinhibitor,簡稱PI),它能與多種絲氨酸蛋白酶結合形成複合體,包括彈性蛋白酶、胰蛋白酶、糜蛋白酶、凝血酶及細菌蛋白酶。其重要的作用在於抑制中性粒細胞的彈性蛋白酶,彈性蛋白酶可作用於肺泡壁的彈性蛋白及其組織的結構蛋白而造成組織損害。因而α1-AT的主要生理功能是保護下呼吸道及其他組織免受彈性蛋白酶的損傷。
2.遺傳學α1-AT的含量是由1組常染色體等位基因的不同類型所決定的,其基因座命名為Pi。用區帶電泳技術方法發現人群血清中有70餘種泳動速度不同的α1-AT帶,等位基因按電泳遷移速度的快慢用英文字母排列如下:B、E、F、G、I、L、M、N、P、O、R、S、V、W、X、Z,經國際會議命名PiM、PiS、Piz等α1-AT的變異型和亞型,組成1個蛋白酶抑制物遺傳表型系統。在遺傳過程中,由兩個M型等位基因組成的純合子PiMM為正常的表現型,PiMM是最常見的正常功能基因型。PiZZ是α1-AT嚴重缺失的等位基因,純合子為ZZ,血清中α1-AT含量是正
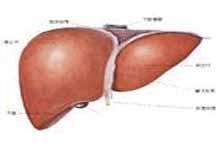 抗胰蛋白酶缺乏症
抗胰蛋白酶缺乏症3.病理生理機制正常情況下體內蛋白酶與抗蛋白酶呈平衡狀態。α1-AT缺乏症中彈性蛋白酶和抗蛋白酶平衡失調,蛋白酶的活性超過蛋白酶抑制過程時,彈性蛋白酶對肺泡結構造成持續損傷,肺組織銷蝕,肺泡間隔破壞,氣腔持久性擴大,臨床上表現為肺氣腫特徵,由於肺泡壁的消失,小氣道在呼氣時失去支架而陷閉,引起肺功能的損害。這主要是損傷了肺泡結締組織中的彈力纖維而致肺氣腫。α1-AT缺乏症是由於肝細胞中合成的α1-AT不能分泌到血漿,而貯積形成包涵體,引起不分泌的原因與Z型蛋白酶聚集有關。其中某些人有發生肝硬化的風險。通過對肝細胞中包涵體分析發現碳水化合物的結構不正常,缺少末端N-乙醯唾液酸殘基,而含有多個甘露糖的核心,是一個加工不完全的糖蛋白。
病理改變
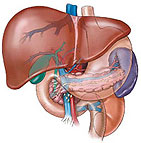 抗胰蛋白酶缺乏症
抗胰蛋白酶缺乏症免疫螢光法和免疫細胞學示沉積包涵體為α1-AT,此包含物在純合子也可看到。此類包涵物的密度和範圍與有無肝病表現似無確切關係,但無該包涵體沉積則無肝病發生,說明該包涵體在該病發病機制中可能起一定作用。
電鏡下可見肝細胞內擴張的粗面內質網內含特徵的形態不一的沉積物,而高爾基體內則無此沉積物,沉積物的量個體差異很大,膽管細胞內也可見到。另外,肝細胞內還可見糖原、空泡、脂褐素及膽汁淤積。
Pizz表型的新生兒,如由於α1-AT缺乏而伴有膽汁鬱積性黃疸,肝臟有如下三種病理表現:
一、肝細胞損傷
特點是肝細胞腫大,相對炎性細胞浸潤輕,且多為單核細胞浸潤,可伴或不伴有肝纖維化,可有淤膽,但門脈區無膽栓形成。
二、門脈性肝纖維化和膽道增生
有廣泛的門脈區纖維化,甚至類似肝硬化的表現,有明顯的膽小管增生,6個月前黃疸消退,但肝、脾進行性腫大、變硬,以後有2例出現了門脈高壓,但該類患者肝外膽管無異常。
三、小膽管發育不良
肝結構正常,有輕微肝細胞損傷,門區僅輕微纖維化,但膽管數明顯減少,膽汁淤積散布在肝小葉內,臨床經過為進行性黃疸,伴有瘙癢和高膽固醇血症。
臨床表現
在出生後第一周可有膽汁淤積性黃疸、大便不著色、尿色深。體檢可發現肝腫大。生化指標有梗阻性黃疸的指征,2~4個月時黃疸往往消失,在2歲以後可現肝硬化。
在成年人,大多數α1-AT缺乏症患者以突出的門脈高壓症為首發表現,患者常死於上消化道出血和/或肝昏迷,常發生肺氣腫。男性肝硬化和肝癌的發病率高於女性。
由α1-AT缺乏導致肝硬化患者,肝臟腫瘤發病率很高,以起源於肝細胞者居多,部分起源於膽管。
診斷檢查
診斷:臨床表現結合病史可作出肺氣腫的診斷,在高加索人群中凡是肺氣腫患者均應除α1-AT缺乏症的可能性。可經血清蛋白電泳、醋酸纖維電泳、放射免疫擴散法和電泳免疫分析等免疫分析,還可測血清胰蛋白酶抑制活性。進一步作定型檢查可幫助確診α1-AT缺乏症。
實驗室檢查:血清蛋白電泳、醋酸纖維電泳、放射免疫擴散法和電泳免疫分析等免疫分析,還可測血清胰蛋白酶抑制活性。
其他輔助檢查:肺殘氣量/肺總量比值增加,呼氣受限,彌散量降低,肺順應性增加。
治療與鑑別
二:
 抗胰蛋白每缺乏
抗胰蛋白每缺乏2.補充治療法輸入適量的α1-AT,使彈性蛋白酶與蛋白酶抑制劑之間達到平衡。一般輸入正常人血漿或血漿製品可補充α1-AT。近年來套用DNA技術,在大腸桿菌和酵母中產生大分子α1-AT用於治療α1-AT缺乏症。
3.人工合成抗蛋白酶製劑的套用人工合成的鹼化劑如氨甲基酮肽,醯化劑如重氨肽類和單肽類均用於α1-AT缺乏者和慢性支氣管炎合併肺氣腫者,但他們的毒性及致癌作用尚不清楚,仍在研究中,一種新型肽類硼酸AAPb在動物試驗中能有效地使整個肺髒不受彈性蛋白酶的損害,這些試驗性治療對因α1-AT缺乏致肺氣腫的防治提供新途徑。
併發症
並發高碳酸血症和呼吸衰竭
預後及預防
預後:吸菸對發病年齡及病程影響很大,不吸菸患者中98%的女性,65%的男性能活至55歲;而吸菸患者中僅30%女性,18%男性能活至同樣年齡。
預防:目前尚無資料。
相關藥品
相關詞條
呼吸內科疾病
| 研究範圍及進展呼吸內科是研究呼吸系統疾病的學科。它是既古老又年輕的學科,說它古老是因為,自從人類認識疾病以來,呼吸系統疾病就一直是危害人類健康的常見病和多發病,八十年代中期的統計資料表明,呼吸系統疾病仍然是導致死亡的主要疾病。在死亡的順序上排列第二。現在我們來認識一下這些可怕的疾病吧? |
