 進行性骨幹發育不良
進行性骨幹發育不良症狀體徵
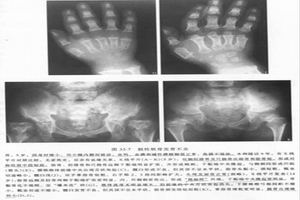 症狀表現
症狀表現Brat等報導1例成年輕型散發性PDD病變累及乾骺端,但無症狀。由於顱底骨硬化常致顱神經孔狹窄,產生顱神經壓迫症狀,加上慢性顱內高壓等原因,可產生聽力減退(80%)、視力障礙、視盤水腫、突眼、復視、面神經麻痹等。偶可引起小腦性共濟失調,病情輕者可無症狀。
本病一般呈進行性發展與惡化,但病程進展速度不一,無自愈可能。deVits等報導1例PDD婦女,妊娠後骨痛、頭痛消失,並可停用糖皮質固醇類藥物,原因未明。據分析,這可能與妊娠時體內某些激素分泌或免疫調節功能變化有關,而分娩後6個月本病又復發加重。
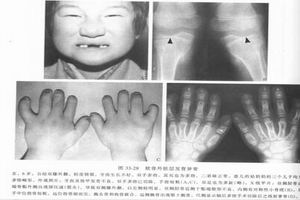
2.體徵體格檢查可見下肢肌肉萎縮,皮下脂肪薄,下肢彎曲畸形,少數有膝外翻,頭顱大,前額突出,性發育遲緩。腰椎過度前突,杵狀指(趾),貧血及智力低下等。個別病人眼底可見視盤水腫,顱內壓增高。有些病人體格無異常,經X線檢查才發現本病。
3.X線檢查
(1)長管骨:骨內、外骨膜骨化而附加於原皮質表層,致使骨皮質增厚、硬化,以骨幹中段顯著,髓腔狹窄或完全消失,但可有斑片狀密度減低區,可見骨周圍軟組織萎縮。受累骨骼的發生頻率依次為脛骨、股骨、肱骨、尺骨、橈骨和腓骨。典型者為對稱分布,通常乾骺受累較輕,偶爾累及骨骺。
(2)短管骨:跖骨常受累,病變較輕,其病變形態與長骨相似,皮質增厚,骨幹增粗,雙側病變基本對稱。
(3)顱骨:顱蓋骨肥厚,主要為內、外板增厚硬化或板障狹窄、消失、顱底骨硬化、顱底神經和血管通過的孔道狹窄。
(4)其他骨骼:個別可見脊柱椎板較緻密,亦可累及肋骨、鎖骨及骨盆。病變的主要表現是皮質骨增厚、硬化。
4.生化檢查
(1)血紅蛋白降低,血沉增快,血鈣、磷正常或低血鈣、高血磷,血PTH、CT正常,血ALP增高,血清骨鈣素(BGP)、Ⅰ型前膠原C端前肽(PICP)增高。
(2)高血磷時,尿磷低、尿羥脯氨酸正常。
(3)少數病人免疫球蛋白A、G、M增高,T淋巴亞群CD4 降低。
本病的生化指標與骨病變有密切關係,測定骨代謝指標可反映骨病變病情,其中Ⅰ型膠原N端前肽(NTX)、Ⅰ型前膠原C端肽(PINP)、骨鈣素(BGP)或骨源性ALP的意義較大。
疾病病因
 疾病病因
疾病病因Makita等報導,PDD的表現型即使在同一家族中也存在明顯的異質性,一家三代的12例患者中,7例的臨床表現典型,另5例僅有骨骼的節段性病變或無症狀性骨幹硬化,這些病人的表現酷似Ribbing病(多發性骨幹硬化症),並認為PDD與Ribbing病是同一骨病的變異表現型。
病理生理
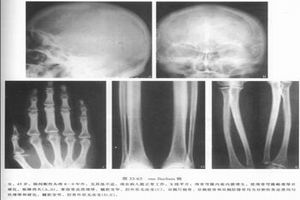 病理生理
病理生理本病變進展緩慢,主要累及四肢長骨,病損自骨幹開始,順長骨之長軸向兩端發展,骨幹膨大,呈梭形,骨皮質增厚,表面不平。骨內外膜下有新骨形成,與骨皮質融合後,皮質骨可呈層狀結構。骨的外層膠原纖維排列紊亂,呈不成熟的交織骨。中層有骨小梁,膠原纖維如網狀形成板層骨。內層為增厚的緻密板層骨。骨小梁粗,排列紊亂,小梁間隙為纖維化的脂肪組織。成骨細胞可增多,活性增強,新骨生成,骨吸收與骨重建緩慢,髓腔變小伴纖維化。
診斷檢查
 診斷檢查
診斷檢查診斷:
1.典型病例有肌無力、骨痛症狀、矮小、消瘦身材。
2.X線示長骨內、外膜增厚,骨髓腔變窄,骨骺及乾骺端、關節面受累等表現。
3.血沉增快、貧血,ALP、骨鈣素、PICP增高,PTHH、CT及尿羥脯氨酸正常等可做出診斷。
實驗室檢查:
1.血紅蛋白降低。
2.血沉增快。
3.血鈣磷、ALP正常。
4.血PTHCT和尿羥脯氨酸亦多正常。
其他輔助檢查:
X線檢查顯示,病變以四肢長骨、顱骨多見,亦可累及掌骨、肋骨、肩胛骨、鎖骨和脊柱。長骨內外膜增厚、骨皮質緻密或呈層狀,骨幹梭形增粗,表面不平,骨髓腔變窄。但病變不累及骨骺、乾骺端及關節面。顱骨內外板增厚,密度增高,有不規則硬化斑,以顱底為明顯。脊椎可有灶性斑片狀硬化影。
鑑別診斷
 形似疾病鑑別
形似疾病鑑別2.石骨症為硬化增生性骨病,主要為長骨骨骺端的軟骨排列紊亂,骨基質膠原成分減少,骨髓腔被骨化,無生化異常,病變常累及脊柱,而PDD雖主要為長管骨病變,但罕有骨骺端受累。骨髓腔變窄是由於骨髓纖維化所致。ALP增高,血沉增快等均可鑑別。
治療方案
 藥物治療
藥物治療2.止痛常用的藥物有以下幾種:
(1)解熱止痛劑:包括非特異性的消炎止痛藥吲哚美辛、布洛芬、吡羅昔康(炎痛喜康)等、水楊酸類(阿司匹林、索米痛片、非那西丁等)。
(2)糖皮質激素:治療PDD的作用有兩個方面:①抑制成骨細胞,減少骨髓腔的狹窄所造成的骨畸形而達到止痛目的。②血清免疫球蛋白及T淋巴細胞亞群CD4 增高,所以疼痛可能有免疫因素的參與。絕大多數病人對糖皮質類固醇類有一定療效。近期報導,有的患者在使用一般藥物或潑尼松治療無效時用地夫可特(deflazacort)1.2mg/(kg?d)12個月後疼痛消失,放射學亦有改善,且無明顯副作用。本藥為類固醇類新藥,其抗炎作用與潑尼松相似,但副作用明顯減少。
3.骨畸形及其他併發症的處理骨畸形嚴重者或影響功能可行截骨矯形術。由於顱神經受壓引起的耳聾,可行耳蝸移植,重建聽覺。由於顱內壓增高,引起的視盤水腫的。
食療方案
 栗子糕
栗子糕乾黃精100克,蜂蜜200克。將洗淨的黃精放入鍋內,加水適量浸泡透發,大火煮滾,改用小火煎煮至熟爛,加蜂蜜再煮滾,調勻即可,待冷裝瓶備用。
黃精補益精氣,強筋健骨。每日食用3次。
2.栗子糕
栗子(板栗)500克,白糖250克。將栗子連殼洗淨,放入鍋中,加水煮半小時,待冷,去殼和皮,放在碗中再上蒸鍋蒸半小時。趁熱取出,放在盆內,加入白糖,用勺壓拌均勻,使成栗茸;填入膏模(或以塑膠瓶蓋、啤酒蓋為模)壓成餅狀。排碟內即可。
栗子健脾益胃,補腎強筋。
預後及預防
預後:治療中由於糖皮質激素免疫抑制作用而止痛,但利用放射學和閃爍掃描發現,可的松及其類似物雖能提高痛閾,減少疼痛,提高病人生活質量,但並不能改善病變進展。預防:因骨痛而制動造成的肌萎縮,除每天堅持按摩、揉捏,活動關節和肌肉外,還可囑其做關節的屈曲運動和肌收縮運動,防止廢用性肌萎縮和營養缺乏造成的機體抵抗力下降。
