
 原子吸收光譜儀
原子吸收光譜儀基本原理
 原子吸收光譜原理圖
原子吸收光譜原理圖每一種元素的原子不僅可以發射一系列特徵譜線,也可以吸收與發射線波長相同的特徵譜線。當光源發射的某一特徵波長的光通過原子蒸氣時,即入射輻射的頻率等於原子中的電子由基態躍遷到較高能態(一般情況下都是第一激發態)所需要的能量頻率時,原子中的外層電子將選擇性地吸收其同種元素所發射的特徵譜線,使入射光減弱。特徵譜線因吸收而減弱的程度稱吸光度A,與被測元素的含量成正比:
 原子中質子,中子及電子-內部結構模型圖
原子中質子,中子及電子-內部結構模型圖 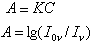
式中K為常數;C為試樣濃度;I0v為原始光源強度;Iv為吸收後特徵譜線的強度。按上式可從所測未知試樣的吸光度,對照著已知濃度的標準系列曲線進行定量分析。
由於原子能級是量子化的,因此,在所有的情況下,原子對輻射的吸收都是有選擇性的。由於各元素的原子結構和外層電子的排布不同,元素從基態躍遷至第一激發態時吸收的能量不同,因而各元素的共振吸收線具有不同的特徵。原子吸收光譜位於光譜的紫外區和可見區。
譜線輪廓
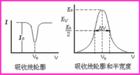 原子吸收光譜曲線
原子吸收光譜曲線原子吸收光譜線並不是嚴格幾何意義上的線,而是占據著有限的相當窄的頻率或波長範圍,即有一定的寬度。原子吸收光譜的輪廓以原子吸收譜線的中心波長和半寬度來表征。中心波長由原子能級決定。半寬度是指在中心波長的地方,極大吸收係數一半處,吸收光譜線輪廓上兩點之間的頻率差或波長差。半寬度受到很多實驗因素的影響。影響原子吸收譜線輪廓的兩個主要因素:
1、都卜勒變寬。都卜勒寬度是由於原子熱運動引起的。從物理學中已知,從一個運動著的原子發出的光,如果運動方向離開觀測者,則在觀測者看來,其頻率較靜止原子所發的光的頻率低;反之,如原子向著觀測者運動,則其頻率較靜止原子發出的光的頻率為高,這就是都卜勒效應。原子吸收分析中,對於火焰和石墨爐原子吸收池,氣態原子處於無序熱運動中,相對於檢測器而言,各發光原子有著不同的運動分量,即使每個原子發出的光是頻率相同的單色光,但檢測器所接受的光則是頻率略有不同的光,於是引起譜線的變寬。
2、碰撞變寬。當原子吸收區的原子濃度足夠高時,碰撞變寬是不可忽略的。因為基態原子是穩定的,其壽命可視為無限長,因此對原子吸收測定所常用的共振吸收線而言,譜線寬度僅與激發態原子的平均壽命有關,平均壽命越長,則譜線寬度越窄。原子之間相互碰撞導致激發態原子平均壽命縮短,引起譜線變寬。碰撞變寬分為兩種,即赫魯茲馬克變寬和洛倫茨變寬。
赫魯茲馬克變寬是指被測元素激發態原子與基態原子相互碰撞引起的變寬,稱為共振變寬,又稱赫魯茲馬克變寬或壓力變寬。在通常的原子吸收測定條件下,被測元素的原子蒸氣壓力很少超過10-3mmHg,共振變寬效應可以不予考慮,而當蒸氣壓力達到0.1mmHg時,共振變寬效應則明顯地表現出來。洛倫茨變寬是指被測元素原子與其它元素的原子相互碰撞引起的變寬,稱為洛倫茨變寬。洛倫茨變寬隨原子區內原子蒸氣壓力增大和溫度升高而增大。
除上述因素外,影響譜線變寬的還有其它一些因素,例如場致變寬、自吸效應等。但在通常的原子吸收分析實驗條件下,吸收線的輪廓主要受都卜勒和洛倫茨變寬的影響。在2000-3000K的溫度範圍內,原子吸收線的寬度約為10-3-10-2nm。
儀器結構
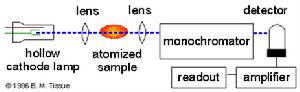
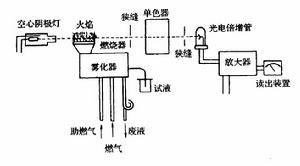 原子吸收光譜儀器結構圖
原子吸收光譜儀器結構圖原子吸收光譜儀由光源、原子化器、分光器、檢測系統等幾部分組成。基本構造右圖
1、 光源。光源的功能是發射被測元素的特徵共振輻射。對光源的基本要求是:發射的共振輻射的半寬度要明顯小於吸收線的半寬度;輻射強度大、背景低,低於特徵共振輻射強度的1%;穩定性好,30分鐘之內漂移不超過1%;噪聲小於0.1%;使用壽命長於5安培小時。 空心陰極放電燈是能滿足上述各項要求的理想的銳線光源,套用最廣。
2、原子化器。其功能是提供能量,使試樣乾燥,蒸發和原子化。 在原子吸收光譜分析中,試樣中被測元素的原子化是整個分析過程的關鍵環節。實現原子化的方法,最常用的有兩種:
火焰原子化法:是原子光譜分析中最早使用的原子化方法,至今仍在廣泛地被套用;
非火焰原子化法,其中套用最廣的是石墨爐電熱原子化法。
3、分光器。它由入射和出射狹縫、反射鏡和色散元件組成,其作用是將所需要的共振吸收線分離出來。分光器的關鍵部件是色散元件,現在商品儀器都是使用光柵。原子吸收光譜儀對分光器的解析度要求不高,曾以能分辨開鎳三線Ni230.003、Ni231.603、Ni231.096nm為標準,後採用Mn279.5和279.8nm代替Ni三線來檢定解析度。光柵放置在原子化器之後,以阻止來自原子化器內的所有不需要的輻射進入檢測器。
4、檢測系統。原子吸收光譜儀中廣泛使用的檢測器是光電倍增管,最近一些儀器也採用CCD作為檢測器。
干擾效應
原子吸收光譜分析中,干擾效應按其性質和產生的原因,可以分為四類:
1、物理干擾
物理干擾是指試樣在轉移、蒸發和原子化過程中,由於試樣任何物理特性(如粘度、表面張力、密度等)的變化而引起的原子吸收強度下降的效應。物理干擾是非選擇性干擾,對試樣各元素的影響基本是相似的。配製與被測試樣相似組成的標準樣品,是消除物理干擾最常用的方法。在不知道試樣組成或無法匹配試樣時,可採用標準加入法或稀釋法來減小和消除物理干擾。
2、化學干擾
化學干擾是由於液相或氣相中被測元素的原子與干擾物質組分之間形成熱力學更穩定的化合物,從而影響被測元素化合物的解離及其原子化。磷酸根對鈣的干擾,矽、鈦形成難解離的氧化物、鎢、硼、希土元素等生成難解離的碳化物,從而使有關元素不能有效原子化,都是化學干擾的例子。化學干擾是一種選擇性干擾。
消除化學干擾的方法有:化學分離;使用高溫火焰;加入釋放劑和保護劑;使用基體改進劑等。例如磷酸根在高溫火焰中就不干擾鈣的測定,加入鍶、鑭或EDTA等都可消除磷酸根對測定鈣的干擾。在石墨爐原子吸收法中,加入基體改進劑,提高被測物質的穩定性或降低被測元素的原子化溫度以消除干擾。例如,汞極易揮發,加入硫化物生成穩定性較高的硫化汞,灰化溫度可提高到300℃;測定海水中Cu、Fe、Mn、As,加入 NH4NO3,使NaCl轉化為NH4Cl,在原子化之前低於500℃的灰化階段除去。
3、電離干擾
在高溫下原子電離,使基態原子的濃度減少,引起原子吸收信號降低,此種干擾稱為電離干擾。電離效應隨溫度升高、電離平衡常數增大而增大,隨被測元素濃度增高而減小。加入更易電離的鹼金屬元素,可以有效地消除電離干擾。
4、 光譜干擾
光譜干擾包括譜線重疊、光譜通帶記憶體在非吸收線、原子化池內的直流發射、分子吸收、光散射等。當採用銳線光源和交流調製技術時,前三種因素一般可以不予考慮,主要考慮分子吸收和光散射的影響,它們是形成光譜背景的主要因素。
特點
主要有以下優點:
1 選擇性強。這是因為原子吸收頻寬很窄的緣故。因此,測定比較快速簡便,並有條件實現自動化操作。在發射光譜分析中,當共存元素的輻射線或分子輻射線不能和待測元素的輻射線相分離時,會引起表觀強度的變化。
而對原子吸收光譜分析來說:譜線干擾的幾率小,由於譜線僅發生在主線系,而且譜線很窄,線重疊幾率較發射光譜要小得多,所以光譜干擾較小。即便是和鄰近線分離得不完全,由於空心陰極燈不發射那種波長的輻射線,所以輻射線干擾少,容易克服。在大多數情況下,共存元素不對原子吸收光譜分析產生干擾。在石墨爐原子吸收法中,有時甚至可以用純標準溶液製作的校正曲線來分析不同試樣。
2、靈敏度高。原子吸收光譜分析法是目前最靈敏的方法之一。火焰原子吸收法的靈敏度是ppm到ppb級,石墨爐原子吸收法絕對靈敏度可達到10-10~10-14克。常規分析中大多數元素均能達到ppm數量級。如果採用特殊手段,例如預富集,還可進行ppb數量級濃度範圍測定。由於該方法的靈敏度高,使分析手續簡化可直接測定,縮短分析周期加快測量進程;由於靈敏度高,需要進樣量少。無火焰原子吸收分析的試樣用量僅需試液5~100?l。固體直接進樣石墨爐原子吸收法僅需0.05~30mg,這對於試樣來源困難的分析是極為有利的。譬如,測定小兒血清中的鉛,取樣只需10?l即可。
3 分析範圍廣。發射光譜分析和元素的激發能有關,故對發射譜線處在短波區域的元素難以進行測定。另外,火焰發射光度分析僅能對元素的一部分加以測定。例如,鈉只有1%左右的原子被激發,其餘的原子則以非激發態存在。
在原子吸收光譜分析中,只要使化合物離解成原子就行了,不必激發,所以測定的是大部分原子。目前套用原子吸收光譜法可測定的元素達73種。就含量而言,既可測定低含量和主量元素,又可測定微量、痕量甚至超痕量元素;就元素的性質而言,既可測定金屬元素、類金屬元素,又可間接測定某些非金屬元素,也可間接測定有機物;就樣品的狀態而言,既可測定液態樣品,也可測定氣態樣品,甚至可以直接測定某些固態樣品,這是其他分析技術所不能及的。
4、抗干擾能力強。第三組分的存在,電漿溫度的變動,對原子發射譜線強度影響比較嚴重。而原子吸收譜線的強度受溫度影響相對說來要小得多。和發射光譜法不同,不是測定相對於背景的信號強度,所以背景影響小。在原子吸收光譜分析中,待測元素只需從它的化合物中離解出來,而不必激發,故化學干擾也比發射光譜法少得多。
5、精密度高。火焰原子吸收法的精密度較好。在日常的一般低含量測定中,精密度為1~3%。如果儀器性能好,採用高精度測量方法,精密度為<1%。無火焰原子吸收法較火焰法的精密度低,目前一般可控制在15%之內。若採用自動進樣技術,則可改善測定的精密度。火焰法:RSD <1%,石墨爐 3~5%。
原子吸收光譜有以下一些不足:
原則上講,不能多元素同時分析。測定元素不同,必須更換光源燈,這是它的不便之處。原子吸收光譜法測定難熔元素的靈敏度還不怎么令人滿意。在可以進行測定的七十多個元素中,比較常用的僅三十多個。當採用將試樣溶液噴霧到火焰的方法實現原子化時,會產生一些變化因素,因此精密度比分光光度法差。現在還不能測定共振線處於真空紫外區域的元素,如磷、硫等。
標準工作曲線的線性範圍窄(一般在一個數量級範圍),這給實際分析工作帶來不便。對於某些基體複雜的樣品分析,尚存某些干擾問題需要解決。在高背景低含量樣品測定任務中,精密度下降。如何進一步提高靈敏度和降低干擾,仍是當前和今後原子吸收光譜分析工作者研究的重要課題。
發展歷史
1、第一階段——原子吸收現象的發現與科學解釋
早在1802年,伍朗斯頓(W.H.Wollaston)在研究太陽連續光譜時,就發現了太陽連續光譜中出現的暗線。1817年,弗勞霍費(J.Fraunhofer)在研究太陽連續光譜時,再次發現了這些暗線,由於當時尚不了解產生這些暗線的原因,於是就將這些暗線稱為弗勞霍費線。1859年,克希荷夫(G.Kirchhoff)與本生(R.Bunson)在研究鹼金屬和鹼土金屬的火焰光譜時,發現鈉蒸氣發出的光通過溫度較低的鈉蒸氣時,會引起鈉光的吸收,並且根據鈉發射線與暗線在光譜中位置相同這一事實,斷定太陽連續光譜中的暗線,正是太陽外圍大氣圈中的鈉原子對太陽光譜中的鈉輻射吸收的結果。
2、第二階段——原子吸收光譜儀器的產生
原子吸收光譜作為一種實用的分析方法是從1955年開始的。這一年澳大利亞的瓦爾西(A.Walsh)發表了他的著名論文“原子吸收光譜在化學分析中的套用”奠定了原子吸收光譜法的基礎。50年代末和60年代初,Hilger, Varian Techtron及Perkin-Elmer公司先後推出了原子吸收光譜商品儀器,發展了瓦爾西的設計思想。到了60年代中期,原子吸收光譜開始進入迅速發展的時期。
3、第三階段——電熱原子吸收光譜儀器的產生
1959年,蘇聯里沃夫發表了電熱原子化技術的第一篇論文。電熱原子吸收光譜法的絕對靈敏度可達到10-12-10-14g,使原子吸收光譜法向前發展了一步。近年來,塞曼效應和自吸效應扣除背景技術的發展,使在很高的的背景下亦可順利地實現原子吸收測定。基體改進技術的套用、平台及探針技術的套用以及在此基礎上發展起來的穩定溫度平台石墨爐技術(STPF)的套用,可以對許多複雜組成的試樣有效地實現原子吸收測定。
4、第四階段——原子吸收分析儀器的發展
隨著原子吸收技術的發展,推動了原子吸收儀器的不斷更新和發展,而其它科學技術進步,為原子吸收儀器的不斷更新和發展提供了技術和物質基礎。近年來,使用連續光源和中階梯光柵,結合使用光導攝象管、二極體陣列多元素分析檢測器,設計出了微機控制的原子吸收分光光度計,為解決多元素同時測定開闢了新的前景。微機控制的原子吸收光譜系統簡化了儀器結構,提高了儀器的自動化程度,改善了測定準確度,使原子吸收光譜法的面貌發生了重大的變化。聯用技術(色譜-原子吸收聯用、流動注射-原子吸收聯用)日益受到人們的重視。色譜-原子吸收聯用,不僅在解決元素的化學形態分析方面,而且在測定有機化合物的複雜混合物方面,都有著重要的用途,是一個很有前途的發展方向。
近年研究展望
 原子吸收光譜
原子吸收光譜近年來國內外都有人致力於研究雷射在原子吸收分析方面的套用:
(1)用可調諧雷射代替空心陰極燈光源。
(2)用雷射使樣品原子化。它將為微區和薄膜分析提供新手段、為難熔元素的原子化提供了新方法。塞曼效應的套用,使得能在很高的背景下也能順利地實現測定。連續光源、中階梯光柵單色器、波長調製原子吸收法(簡稱CEWM-AA法)是70年代後期發展起來的一種背景校正新技術。它的主要優點是僅用一個連續光源能在紫外區到可見區全波段工作,具有二維空間色散能力的高分辨本領的中階梯光柵單色器將光譜線在二維空間色散,不僅能扣除散射光和分子吸收光譜帶背景,而且還能校正與分折線直接重疊的其他原子吸收線的干擾。使用電視型光電器件做多元素分析鑑定器,結合中階梯光柵單色器和可調諧雷射器代替元素空心陰極燈光源,設計出用電子計算機控制的測定多元素的原子吸收分光光度計,將為解決同時測定多元素問題開闢新的途徑。高效分離技術氣相色譜、液相色譜的引入,實現分離儀器和測定儀器聯用,將會使原子吸收分光光度法的面貌發生重大變化,微量進樣技術和固體直接原子吸收分析受到了人們的注意。固體直接原子吸收分析的顯著優點是:省去了分解試樣步驟,不加試劑,不經任何分離、富集手續,減少了污染和損失的可能性,這對生物、醫藥、環境、化學等這類只有少量樣品供分析的領域將是特別有意義的。所有這些新的發展動向,都很值得引起我們的重視。近年來,微型電子計算機套用到原子吸收分光光度計後,使儀器的整機性能和自動化程度達到一個新的階段。
目前原子吸收法已廣泛套用於各個領域,對工業、農業、醫藥衛生、教學科研等發展起著積極的作用
的的化學通用分析儀器
| 化學通用分析儀器主要用於中間體、染料、醫藥、生化、環保、石油化工、食品、農藥等各類在紫外-可見光區有一定吸收的精細化工產品的分離分析。 |
