疾病概述
 病毒傳播
病毒傳播其病理生理改變大致可以分為3類:
①通過該代謝途徑的某些終末產物缺乏,如過氧化酶體病、溶酶體病等,產生的症狀多為持續性、進行性的,且與進食等因素無關;
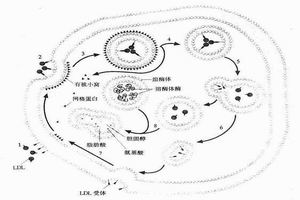 遺傳性代謝缺陷病
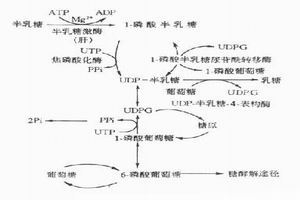
遺傳性代謝缺陷病③由於代謝途徑受阻而導致對肝、腦、肌等組織的供能不足,如糖代謝障礙。先天性高乳酸血症、脂肪酸氧化缺陷、線粒體呼吸鏈功能障礙等,臨床常見低血糖、高乳酸血症、肌張力低下、體重不增等表現。
症狀體徵
 傳播方式
傳播方式【神經系統症狀】
神經系統症狀是先天性代謝缺陷在新生兒期最先呈現的常見症狀。不論是何種原發疾病,由中間或(和)旁路代謝產物累積的毒性作用所致者,患兒最先常表現為吸吮和餵養困難,繼而出現呼吸異常或暫停、呃逆、心律緩慢、體溫不升等,甚至進入昏迷;患兒肌張力常陣發增高,重症且可見角弓反張,或伴有拳擊狀或蹬踏狀的肢體慢動作,亦有呈現軀幹肌張力降低而肢體肌張力增高且伴有震顫或肌陣攣者,易被誤認為驚厥。由於代謝障礙而致供能缺陷者則發病較早,神經系統症狀輕重不一,以肌張力低下為主,但甚少嗜睡和昏迷,且大多呈現高乳酸血症和代謝性酸中毒。
 病毒分子
病毒分子肌張力低下是新生兒期最常見的症狀之一,多數是由於缺氧缺血性腦病和重症感染等非遺傳性疾病所造成;或是由於非代謝性的遺傳病,如遺傳性神經肌肉病變和染色體畸變等所導致。少數先天性代謝缺陷病如先天性高乳酸血症、尿素循環酶缺陷、呼吸鏈功能障礙、NKH和過氧化酶體病等,亦常在嗜唾、昏迷、驚厥發作等情況時伴有肌張力降低。
【消化系統症狀】
拒食、嘔吐、腹瀉等頗為常見,這些症狀常在進食後不久發生;持續黃疸伴生長遲緩者常見於crigler-Najjar綜合徵、α1-抗胰蛋白酶缺陷、過氧化酶體病、膽汁酸代謝障礙、C型Niemann-Pick病(慢性神經型)、Byler病等;脂肪酸氧化障礙和尿素循環酶缺陷者可呈現Reye綜合症樣症狀;肝腫大伴有低血糖和驚厥發作者常提示(Ⅰ或Ⅲ型)糖原累積病和高胰島素血症等;肝功能衰竭症狀(黃疸、出血症狀、轉氨酶增高、腹水等)出現時應考慮半乳糖血症、Ⅰ型酪氨酸血症、果糖不耐症和呼吸鏈功能障礙等疾病;各種原因所造成的肝細胞功能衰竭時都可在臨床上發生糖尿、低血糖、高氨血症、高乳酸血症、高酪氨酸血症、高甲硫氨酸血症等情況,必須注意鑑別。
【循環系統症狀】
呼吸鏈功能障礙、脂肪酸氧化障礙、Ⅱ型糖原累積病(Pompe病)可發生心功能衰竭症狀;心肌病變和心律異常尤多見於長鏈脂肪酸氧化障礙患兒。
病理生理
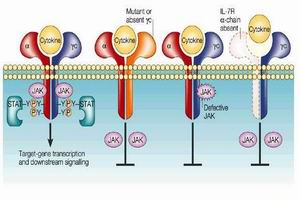 遺傳性代謝缺陷病
遺傳性代謝缺陷病②受累代謝途徑的中間和(或)旁路代謝產物大量蓄積,如苯丙酮尿症、甲基丙二酸尿症、同型胱氨酸尿症、楓糖尿症、半乳糖血症等,通常都呈現累積物導致的中毒症狀,常見者如嘔吐、嗜睡、昏迷、生長發育遲滯、低血糖、高氨血症和酮、酸中毒等,其發病或早或遲,發病前常有無症狀期,或症狀呈間隙發作;
③由於代謝途徑受阻而導致對肝、腦、肌等組織的供能不足,如糖代謝障礙。先天性高乳酸血症、脂肪酸氧化缺陷、線粒體呼吸鏈功能障礙等,臨床常見低血糖、高乳酸血症、肌張力低下、體重不增等表現。
上述病理生理變化多數會直接或間接地影響器官,特別是腦的發育和功能,導致殘疾,甚或危及生命。這類遺傳性疾病在1908年即被Garrod稱之為先天性代謝缺陷,都是單基因疾病,絕大多數屬常染色體隱性遺傳。這類疾病雖然罕見,但通常對機體造成的損害很大,尤其是在早期即累及神經系統,預後甚差。因此,臨床醫師對此必須提高警惕,致力於早期診斷,減少漏診、誤診,及早干預以避免傷殘發生。確切的診斷不僅對先證患兒的治療有利,亦有益於遺傳諮詢、改善人口素質。
診斷檢查
 診斷
診斷2.體檢除注意一般體格發育外,重點需作皮膚、肌肉、骨骼、肝脾、心臟、神經系統的詳細檢查。特別注意眼科情況,有無白內障、角膜色素沉著、虹膜缺損,眼底檢查注意有無黃斑色素沉著、視神經萎縮等。
3.檢驗①初步過篩試驗:根據臨床表現查血中電解質、pH、血氨、尿酸、乳酸、血脂、膽紅素、蛋白質、血糖,尿中還原物質、胺基酸、酮體、乳酸等。X線、B超、腦電圖、心電圖、CT、磁共振等檢查視情況進行,作出初步診斷。②進一步診斷:根據初篩結果,選擇測定血中蓄積物質含量及尿中排泄物質情況,如血中半乳糖、果糖、胺基酸、有機酸、銅、銅藍蛋白、TSH、rT3、皮質激素,尿中乳糖、胺基酸、尿酸及其他有機酸含量,測定代謝異常情況,如葡萄糖耐量試驗、半乳糖耐量試驗、白細胞內包涵體等。在一般情況下,對常見代謝缺陷病可以作出診斷。③明確診斷:測定特殊酶的活性,根據酶含量減少的情況,明確診斷為何種代謝缺陷病。
治療方案
 藥物治療
藥物治療(1)禁其所忌:早期控制飲食,對一些代謝缺陷病有明顯療效,可阻止病情發展。①苯丙酮尿症:從生後2個月開始給低苯丙氨酸飲食,代以水解蛋白,直至6歲左右。②半乳糖血症:從新生兒開始不餵乳類及含半乳糖食物,代以穀類、水果、代乳粉、肉、蛋類飲食。
(2)去其所余:用藥物將體內過多蓄積物排出體外。①肝豆狀核變性,可用絡合劑D-青黴胺20mg/(kg?d)絡合體內過多的銅。②原發性痛風:可用丙磺舒等藥,既減少腎小管對尿酸的重吸收,又使尿酸排出增多。
(3)補其所缺:補充體內缺乏物質。①血友病:給患者補充抗血友病球蛋白、新鮮全血、新鮮血漿。②抗維生素D性佝僂病:口服中性磷酸鹽(磷酸二氫鈉18g及磷酸氫二鈉145g,加水至1000ml,10~20ml,5/d)。同時口服維生素D,1萬~5萬IU/d,最大10萬IU/d,或雙氫速變固醇(DHT),可達2mg/d,2~4周后改為0.5~1mg/d,分次服。也可口服1,25(OH)2D3或25(OH)D31~2μg/d。③酶療法:採取誘導或補充所缺酶的方法治療。酶誘導:Crigler-Najjar綜合徵和Gilbert綜合徵都是由於葡萄糖醛醯轉移酶缺乏,使間接膽紅素不能轉化為直接膽紅素而發生黃疸。可用苯巴比妥、可拉明等酶誘導劑。酶補充:如糖原積累症I型可補充。α-葡萄糖苷酶(黑麴黴菌中提取),高雪病補充葡萄糖苷酶(牛脾中提取)。從人尿中提取芳基硫酸脂酶A治療異染性腦白質營養不良等。
2.器官移植可用胎肝、胎腦、腎臟、骨髓、肝臟等移植治療遺傳代謝性疾病。
3.對症治療有癲癎者抗癲癎治療,骨骼畸形者手術矯正,白內障作手術摘除,脾大者切脾。
4.基因治療將外源正常基因轉入體內,以替代突變或缺陷基因,並在體內長期表達治療疾病。如半乳糖血症植入。α-1-磷酸半乳糖尿甙轉移酶基因,精氨酸血症植入精氨酸分解酶基因等。但目前尚不能作為常規套用。
保健貼士
 預防措施
預防措施1、半乳糖血症:從新生兒開始不餵乳類及含半乳糖的食物,如豆漿、食糖等,代以穀類、水果、肉、蛋類食物。
2、苯丙酮酸尿症:從出生後2個月開始,給予低苯丙酮酸飲食,代以水解蛋白,直至6歲左右。
3、肝豆狀核變性:必須限制含銅飲食。藥物治療:補充缺乏的酶、輔酶或代謝物質,排除多餘物質。
4、抗維生素D性佝僂病,給予維生素D治療,並定期檢查,觀察療效。
5、肝豆狀核變性,給予二巰基丙醇、D—青黴胺治療。
流行病學
 流行病學
流行病學在中國每年2000多萬的出生人口中,約有40萬到50萬的兒童患有遺傳代謝病。它給患兒家庭和社會帶來了巨大的危害。
對遺傳代謝病的患者而言,愈早發現,愈早治療,對患兒愈好。新生兒疾病篩查是提高出生人口素質、減少出生缺陷的第三級預防措施,是嬰兒邁入健康人生的第一道“安檢”。國家《母嬰保健法》及其實施辦法已明確規定醫療保健機構應逐步開展新生兒疾病篩查,並將其列入母嬰保健技術服務項目。李教授強調:“遺傳代謝病是新生兒疾病篩查中重點篩查的一大類疾病,進行更多病種、更高覆蓋率的篩查對降低出生缺陷、提高人口素質意義重大。”
