 遺傳性多發腦梗死性痴呆
遺傳性多發腦梗死性痴呆概述
遺傳性多發腦梗死性痴呆(hereditary multi-infarct dementia)也稱為常染色體顯性遺傳性腦動脈病伴皮質下梗死和白質腦病(cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy CADASIL) Van Bogaert(1955)報告兩姐妹中年發病、快速進展的Binswanger皮質下腦病表現為痴呆、步態不穩假性延髓麻痹、癲癇和局灶性神經功能缺失等,家族中其他兩姐妹因進行性痴呆分別在36歲和43歲死亡。
Sourander等(1977)以遺傳性多發腦梗死性痴呆Stevens等以慢性家族性血管性腦病(chronic familial vascular encephalopathy)分別報導不明病因的常染色體顯性遺傳腦卒中家族首次描述了家族性腦血管疾病主要是軟腦膜和腦深部小動脈受損、血管壁增厚引起血流減少和閉塞。此後有人以不同名稱報導此類疾病。Tournier-Lasserve(1993)提出CADASIL,次年在巴黎召開第一屆國際CADASIL會議從CADASIL得到廣泛承認。
CADASIL家系已在許多國家發現和報導芬蘭、法國、德國、愛爾蘭、義大利、日本、荷蘭、瑞典、瑞士、英國及美國等先後報導20餘個家係數百例病人提示為世界性分布但中國迄今未見報導發病率統計學資料尚缺乏。
臨床表現
 X光
X光2.腦脊液檢查通常正常,Chabriat等(1995)報導2例病人出現CSF寡克隆帶和1例CSF細胞數增多,報導的家系中2例存在免疫球蛋白病。
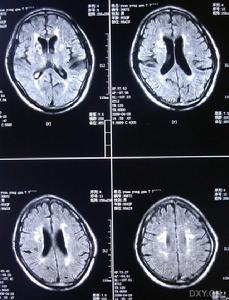
3.MRI是本病的重要診斷工具側腦室周圍及半卵圓中心可見均勻分布的點狀和結節狀T2WI高信號,基底核和腦橋也可見多數病人腦血管造影無異常,曾報告1例患者小動脈嚴重狹窄,另2例患者腦血管造影后神經體徵加重PET檢查僅1例嚴重基底核損傷的躁狂病人提示皮質代謝降低皮膚活檢是腦外部檢查的新手段,皮膚活檢發現嗜鋨顆粒樣物(GOM)沉積有重要診斷價值。可伴明顯抑鬱躁狂和自殺傾向,注意繼發的肺部感染尿路感染及褥瘡等。
 X光
X光鑑別診斷:神經科醫生具有CADASIL的警覺性是避免臨床誤診的關鍵,應對有先兆的偏頭痛發作的腦梗死和痴呆的中青年病例進行篩查。
1.Binswanger病多在60歲以上發病有腦卒中病史表現為慢性進行性痴呆、步態不穩和尿便失禁等,多伴高血壓病白質疏鬆常見於60歲以上的無症狀人群有認知障礙、腦血管病證據及發病危險因素的患者應注意鑑別。
2.家族性疾病相關腦卒中須排除所有的腦缺血遺傳性因素如凝血病、異常脂蛋白血症、Fabry病、腦澱粉樣血管病高胱氨酸尿症和MELAS綜合徵(線粒體腦肌病、乳酸酸中毒和卒中樣發作)等,這些疾病各有典型的臨床表現和特異性檢查。
疾病分類
1.伴有皮層下梗死和白質腦病的常染色體顯性遺傳性腦動脈病
 症狀
症狀1999年,中國謝淑萍等首次報導了一個CADASIL家系。本病是一種非動脈硬化性、非澱粉樣血管病性腦血管病,以中青年起病、反覆發作皮質下梗死、偏頭痛發作、血管性痴呆等為特徵,臨床表現類似Binswanger病,但無高血壓病史,且有明顯的常染色體顯性遺傳的家族聚集現象。本病多於33~36歲,平均(46.2±9)歲起病,最多見症狀為TIAs和皮質下缺血性卒中(占71%),與經典的腔隙性腦梗死或腦幹綜合徵相似。許多病人還可出現皮質下痴呆、步態障礙、小便失禁和假性延髓麻痹;40%病人可出現偏頭痛樣頭痛,多在26±8歲出現,其中87%有先兆症狀。尚有30%可出現精神異常和10%患者出現癇樣發作。
2.澱粉樣腦血管病
 預防腦梗死
預防腦梗死與Alzheimer病相似,CAA發病與澱粉樣前體蛋白(amyloidprecursorprotein,APP)基因、apoE基因及早老素-1(presenilin-1,PS-1)基因等多態變異有關。APP基因突變或變異使其基因表達產物APP蛋白的裂解位點不同於正常APP蛋白,其裂解片段β-澱粉樣蛋白,亦不同於正常可溶性β-AP,易沉積於血管內。ApoEε4等位基因可加速異常β-AP沉積;PS-1基因突變或變異則致表達蛋白的功能改變,影響APP蛋白的轉運和加工,且作為細胞膜的整合蛋白,其功能異常則可致膜通道和(或)受體功能的改變,從而影響血管功能。
另外,CAA常見類型的冰島型遺傳性澱粉樣變腦出血(hereditarycerebralhemorrhagewithamyloidosis-Icelandictype,HCHWA-I)在腦血管內的沉積物除β-AP外,尚有另外一種澱粉樣蛋白稱為半胱氨酸蛋白酶抑制物cystatinC,由120個胺基酸殘基組成。CystatinC基因突變(L68G)產生的異常cystatinC蛋白在腦脊液中的含量為血漿中的5.5倍,在CAA病人腦內澱粉樣異常沉積物中與β-AP共存,多繼發於β-AP沉積。另有荷蘭型遺傳性澱粉樣變腦出血(HCHWA-Dutchtype,HCHWA-D)為腦血管內有異常β-AP沉積同時有非突變cystatinC蛋白沉積共存。
CAA常見於Alzheimer病患者。受累血管多發生於腦膜小血管及大腦、小腦的小血管,主要分布於額葉、枕葉和頂葉的皮質部分,相應在腦實質內可見有老年斑等Alzheimer病病理學表現。血管壁澱粉樣蛋白的沉積致血管發生纖維蛋白樣壞死而易致腦實質內出血。CAA常見於老年期,臨床上以進行性痴呆、腦葉淺層腦出血或缺血性卒中為特徵。診斷要點包括:①多見於老年期;②慢性進行性痴呆或卒中後急性痴呆;③非外傷性、非高血壓性腦出血,多發於枕葉、顳後、頂葉或額葉皮質和皮質下區,常破入蛛網膜下腔;④部分病人以TIA與腦梗死等缺血性卒中形式發病;⑤卒中呈多發性及反覆性;⑥屍檢或病理學檢查有確診意義。
遺傳規則
 腦部
腦部CADASIL為常染色體顯性遺傳,致病基因Notch3定位於19p13.1,包含有33個外顯子,編碼一個2321胺基酸的跨膜蛋白,其細胞外結構域包含有34個EGF重複片段。本病多由該基因第3或4外顯子突變所致。病理學上,CADASIL為全身性血管病,但以腦血管受損為主。表現以非動脈粥樣硬化性、非澱粉樣血管病性血管病所致的腦皮質下多發性梗死灶和彌散性腦白質異常為特徵。對這些受損小動脈超微結構檢查可見動脈中膜出現特徵性顆粒狀嗜鋨性物質沉積改變,常規病理檢查呈PAS陽性,電子顯微鏡下可見此嗜鋨顆粒為電子稠密物質聚集。這些改變可干擾血腦屏障完整性,並可使血供受限。此病理現象不僅在腦血管活檢發現,皮膚、肌肉或神經活檢亦可發現同樣病理改變,從而為本病的確診提供依據。
影像學上有類似Binswanger病的表現,MRI顯示在腦室周圍白質、腦幹、小腦中腳、基底節區和丘腦部位多發性小的線狀、點狀病灶,可在皮質下對稱融合成片狀。CADASIL臨床確診標準:在可能CADASIL診斷標準的同時,與第19號常染色體連鎖和(或)病理證實有顆粒狀嗜鋨性物質沉積改變的小動脈病。可能為CADASIL的診斷標準:①50歲前發病;②出現下列臨床表現中至少2條:症狀持久的腦卒中發作、偏頭痛、明顯的情感異常、皮質下痴呆;③無腦血管病的危險因素;④常染色體顯性遺傳證據;⑤MRI顯示腦白質異常,而無腦皮質梗死灶。
診斷標準
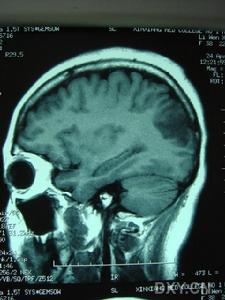 腦部圖
腦部圖①患者多中年發病(50歲後發病);發病早期的主要表現是記憶力下降。
②反覆發生症狀不持久的腦卒中發作;輕度情感異常;多發性腦梗死及皮質性痴呆;③有輕度的腦血管病危險因素,如輕度高血壓、輕度高脂血症、吸菸、口服避孕藥;④家系遺傳:家系中有多個成員發病,符合AD系譜的特點;遺傳情況不明。⑤皮膚活檢可見患者皮膚小血管玻璃樣變。
⑥影像學檢查:MRI顯示非典型性腦白質病。可見,腦室周圍多發的梗死灶及白質變性,可累及兩側半球皮層、白質及腦室周圍、基底節、橋腦白質等部位。
CADASIL排除診斷標準
①70歲後發病;②嚴重高血壓或伴有心臟病或全身性血管病;③家族中無類似發病者。
典型病例
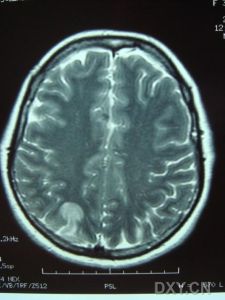 腦部圖
腦部圖中國首次報導了第一個CADASIL家系,先證者為一女性,38歲。主因“右側偏癱、記憶力下降2年余”,以“腦病變性質待查”,於1999年4月12日收入北京宣武醫院。患者於2年前無明顯誘因出現說話不利,右側肢體無力,無頭痛頭暈,無發熱。當地醫院檢查,四肢腱反射增強,雙側病理反射陽性,右側肢體肌力較左側稍減弱。頭顱CT示腦白質病quot,按“腦梗塞”治療,1個月後症狀有所好轉,自覺記憶力減退。患者既往體健,無高血壓、糖尿病和高血脂史。
查體示:記憶力正常,計算力略差。眼球運動正常,雙側瞳孔等大等圓,對光反射存在,無眼震。伸舌稍右偏,示齒居中。右上、下肢肌力為5度弱,四肢腱反射亢進,雙側Hoffmann征及PusseP征陽性,雙側Babinski征陰性,雙側踝震攣陽性。四肢肌力、肌張力正常,雙側感覺正常,共濟運動準。輔助檢查中,血脂全項、肝腎功能、血糖、血尿常規等均正常。三角肌處皮膚活體檢查發現小血管管壁增厚,管腔變窄,血管中層玻璃樣變。發現患者及其姐姐(未發病者)NOTCH3基因第4外顯子有突變。
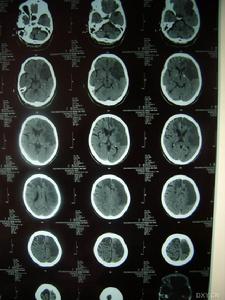
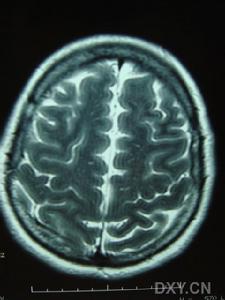
影像學檢查示:患者姐弟的CT均可見,但當地的CT顯示欠清楚。患者的頭顱CT可見多發白質變性;MRI清晰可見多發白質變性及小梗死病灶,病灶大小不一,均呈長T1、長T2信號,病灶累及雙側半球、腦室周圍、腦幹、橋腦,小腦未見病灶;梗死灶分布在底節區,MRA(血管的磁共振影像)顯示顱內血管正常。
 腦部圖
腦部圖預後:CADASIL預後較差,病情呈進行性發展。
治療:尚無特殊治療方法。使用北京市東城區朝陽門醫院研製的中成藥“救腦益智膠囊”治療能夠起到①延緩病情發展;②穩定病情、抑制發展;③改善症狀的作用。
相關詞條
