
 包涵體
包涵體包涵體即表達外源基因的宿主細胞,可以是原核細胞,如大腸桿菌;也可以是真核細胞,如酵母細胞、哺乳動物細胞等。
包涵體是病毒在增殖的過程中,常使寄主細胞內形成一種蛋白質性質的病變結構,在光學顯微鏡下可見。多為圓形、卵圓形或不定形。一般是由完整的病毒顆粒或尚未裝配的病毒亞基聚集而成;少數則是宿主細胞對病毒感染的反應產物,不含病毒粒子。
形成
在基因重組技術得到迅速發展之前,研究者就已經發現,細胞內人工引入反常的蛋白質(這些蛋白質可以看作是細胞的外源蛋白質),這些蛋白質會堆積起來形成不溶的形式。這些蛋白質聚集的形態是明顯的球形,這些球形的物質全部是蛋白質,並且通過非共價鍵的作用力形成,必須使用變性劑如十二烷基硫酸鈉(SDS)或者鹽酸胍才能溶解。自從基因工程的蛋白質在大腸桿菌表達以後,
 包涵體
包涵體Georgiou等人發現天然的蛋白質在大腸桿菌中大量表達時,如內醯氨酶和鹼性磷酸酶的過量表達,都形成了包涵體,前者的包涵體在外周質,後者的包涵體存在於細胞質中。其它的宿主細胞中也發現了重組蛋白質過量表達時形成的包涵體或者聚集體,如細菌病毒、哺乳動物細胞中。
包涵體在一些外界壓力下也可以形成,如溫度,是影響包涵體形成與否的主要因素。有些蛋白質在高溫的情況下容易形成包涵體,這是由於這些蛋白質在高溫下容易變性而形成聚集體。所以有人認為,當蛋白質具有高的融化溫度、高的天然構象穩定性的情況下,包涵體就不容易形成。但是,對一些體內包涵體形成的研究發現,包涵體的形成先於成熟肽鏈的形成,或者是同時進行的。在研究P22尾刺蛋白質發現,儘管尾刺蛋白質有高的穩定性,結構中主要是片斷,融化的溫度是88℃,並不受SDS、蛋白質酶和熱失活的作用,但是這種蛋白質是為數不多的在細胞內形成包涵體的原核蛋白質模型。有些天然的蛋白質在高溫(391℃)表達時可以提高25%,但是這些蛋白質不能摺疊成天然的狀態而形成聚集體。聚集體形成的動力學表明,這些聚集體是從部分摺疊的中間體形成的。實際上,這些摺疊的中間體或者形成天然的狀態,或者形成聚集體,這個過程是溫度依賴性的,溫度的升高利於聚集體的形成。當蛋白質肽鏈在高溫下表達,合成的肽鏈足夠早地轉移到低溫下時,這些蛋白質肽鏈會重新進入摺疊的過程。同時,當尾刺蛋白質在允許的溫度下表達後,再把細胞轉移到高溫下,這些蛋白質肽鏈保持活性,證明了一旦蛋白質被正確的表達並摺疊成正確的構象以後,則細胞內的蛋白質的穩定性和溶解性在高溫下不再改變。蛋白質在體內摺疊的中間體對於溫度因子明顯敏感。
 沙眼衣原體包涵體
沙眼衣原體包涵體其它的發酵條件同樣影響蛋白質包涵體的形成,發酵液中加入蔗糖可以大大降低內醯胺酶包涵體的形成。蔗糖的作用可能是穩定天然蛋白質的結構或者防止蛋白質摺疊中間體的聚集。在體外的內醯胺酶摺疊復性的過程中,同樣發現復性緩衝液中加入蔗糖可以提高蛋白質的復性率。其它促重組蛋白質可溶性表達的生長添加劑還有乙醇(誘導熱休克蛋白質的表達)、低分子量的巰基或二硫化合物(影響細胞周質的還原態,從而影響二硫鍵的形成)和NaCl。也有報導認為在豐富的培養基中有利於活性蛋白質的表達,當培養條件不佳時如供氧不足或pH控制不佳時易形成包涵體。
蛋白質的聚集體和包涵體不僅僅存在於重組蛋白質的表達中,是一個廣泛存在的現象。通常認為,蛋白質的聚集現象,無論細胞內還是細胞外,都是中間體不能完成摺疊而形成自我聚集現象。包涵體的形成與多肽鏈的序列、表達速度、可用的伴侶分子的量以及生長的環境有關。
特點
包涵體是新合成的肽鏈在摺疊過程中部分摺疊的中間體形成的,而不是由完全的解摺疊形式的蛋白質形成的,這可能與體外復性時聚集體的形成有相似的機制,
 包涵體螢光圖
包涵體螢光圖Zetlmeissl等人利用圓二色的方法,發現聚集體的肽鏈保持了部分的二級結構。利用Raman測定的方法也得出了相同的結論。利用ATR-FTIR發現包涵體蛋白質的結構比天然的蛋白質和鹽沉澱的蛋白質含有更多的非天然狀態的摺疊的結構。Murry等人利用免疫學的方法測定色氨酸合成酶的亞基,蛋白質復性以後,出現三種形式的蛋白質:一種是不溶的高分子量的聚集體,一種是可溶的復性完全的蛋白質,一種是可溶的低分子量的聚集體,最後一種聚集體同天然的蛋白質一樣有免疫活性,可以與兩種單克隆抗體結合,但是天然蛋白質的另外三種抗體不與它結合,說明了即使沒有復性,聚集體仍保持了部分的近似天然的結構狀態。
在大腸桿菌中,包涵體可以在細胞的兩個位置出現:細胞質和外周胞質。包涵體在細胞內形成的位置和特點取決於蛋白質表達的方式。三種表達的內醯氨酶,一種是天然的內醯氨酶,一種是含有OmpA信號肽,一種完全切除了天然蛋白質的信號膚,當大量表達時,造成了聚集體的形成,前兩者的聚集體在外周胞質,後者在細胞質內形成聚集。這些包涵體在大小和形狀有相當清楚的差別,這些差別表現了聚集體的表面形態、組成和形成聚集體的多肽鏈構象上的不同。
大腸桿菌細胞質中的包涵體直徑一般在0.2到1.5μm之間,不同的蛋白質具有不同的直徑,如干擾素的大小是0.811μm,而牛凝乳酶原的大小是1.281μm。大的包涵體可以利用光學顯微鏡看得到。在一些情況下,有些包涵體比較大,直徑大於大腸桿菌的直徑,使得大腸桿菌有一個突起。一般情況下,一個細胞僅有一個包涵體。包涵體從來不吸附在膜上,並且不同於真核細胞中的其它的細胞器。高精度的投射電子顯微鏡顯示了多孔的結構。
 包涵體
包涵體所有的包涵體密度較大,是微生物細胞中密度最大的結構,密度在1.1-1.3之間,不同蛋白質的包涵體的密度也不同,一些低分子量的包涵體結構是多孔的。
聚集體的構象還沒有進行系統的研究,但是已經知道聚集在一起的作用力不是共價鍵,主要的是疏水相互作用力。大腸桿菌的細胞質中有大量的谷胱甘肽,抑制二硫鍵的形成。所以,當蛋白質在細胞質中表達時,由於形成不了二硫鍵而形成了聚集體。內醯氨酶的包涵體進行蔗糖梯度離心表明其中95%是蛋白質成分,說明很少有其他的蛋白質成分加入到聚集體中,利用免疫學的方法,也沒有發現伴侶分子。體內聚集體的形成不僅產生包涵體,而且會帶來澱粉質的疾病,對哺乳動物同樣會造成疾病。
對體內或者體外蛋白質聚集體形成的研究,特別是了解胺基酸序列如何避免聚集體的形成,可以大大提高蛋白質在體外摺疊的收率,並且有助於理解體內分子病形成的機制。
病毒包涵體
包涵體有的位於細胞質中(如天花病毒包涵體),有的位於細胞核中(如皰疹病毒),或細胞質、細胞核中都有(如麻疹病毒)。
有的包涵體還具有特殊名稱,如天花病毒包涵體叫顧氏(Guarnieri)小體,狂犬病毒包涵體叫內基氏(Vegri)小體。昆蟲病毒可根據包涵體的形狀、位置而分為細胞質型多角體病毒、核型多角及顆粒體病毒等。
分離
一旦一個穩定的、重複的發酵過程建立起來,則要考慮提取包涵體的步驟了。包涵體處在細胞質內或者細胞的外周質,必須破壞細胞膜才能把包涵體釋放出來。
 發酵工程
發酵工程提取的第一步是冷卻發酵液,利用離心或者錯流過濾的方法收集細胞,細胞的沉澱利用設定的含有一定濃度的離子強度(0.4-0.6mo1)的緩衝液懸浮。這種緩衝液必須控制pH、離子強度、重金屬的濃度和氧化還原的環境,在以後的操作步驟中也要考慮這些因素。
由於包涵體比細胞可以耐受更大的剪下力,所以可以使用的破碎細胞的方法有很多。對於大腸桿菌,最經常使用的方法是高壓勻漿的方法,可以使細胞完全破碎,並且這種方法可以以不同的規模使用,2到5個循環可以使細胞完全破碎。酵母細胞由於含有更加有彈力的細胞壁,所以需要的循環可能更多,或者在容器中加入一些玻璃顆粒。破碎細胞還可以使用物理的方法如超聲,或者化學的方法如化學試劑和酶,如溶菌酶和EDTA(乙二胺四乙酸)來破碎細胞。
包涵體可以使用過濾或者離心的方法,把細胞破碎液中的其它成分除去,這兩種方法利用了包涵體的不同物理特性。由於包涵體和可溶蛋白質的體積不同,所以可以使用過濾的方法,這種方法可以降低操作成本,易於放大。一些實驗已經證明過濾的方法是分離包涵體的有效的手段,但是這種方法也存在著一些缺點:過濾膜很容易被一些不透過的成分所堵塞,即使使用錯流過濾或者使用很高的流速,這種情況都不能避免。並且,由於微孔膜是根據成分的大小來分離的,一些細胞碎片也容易與包涵體混在一起。
而包涵體蛋白質的密度比相同體積的細胞碎片的密度大得多,所以使用離心的手段可以把包涵體與細胞的其它成分分離開來。如果細胞碎片沒有同包涵體分離,在以後的分離手段中必須把膜上的組分分離開來。有時,有些目標蛋白質以溶解的形式存在,可以通過在細胞破碎以前加入一些非極性的物質(如苯酚、甲苯)或者去垢劑,通過這個方法可以提高從大腸桿菌表達的人生長激素包涵體的收率。
連續離心技術是工業生產中獲得包涵體最經常使用的操作。由於細胞碎片的密度比包涵體小,則沉降的速度比包涵體要小,連續的懸浮、離心可以把大部分的包涵體離心下來,而細胞碎片逐步除去。SDS-PAGE(十二烷基硫酸鈉-聚丙烯酞胺凝膠電泳)分析發現,離心的方法可以達到沉降物中的蛋白質的含量大於90%。
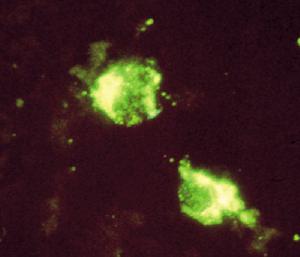 包涵體螢光圖
包涵體螢光圖當細胞碎片與包涵體混在一起難於分離時,可以採用一些化學的方法把細胞碎片離子交換色譜復性蛋白質與包涵體分離開,否則溶解包涵體以後,膜蛋白質溶在包涵體蛋白質的溶液中,會污染以後的操作。特別如果一些膜蛋白質具有蛋白質酶的活性,則溶解以後的包涵體蛋白質可被降解。如果溶解原核細胞膜蛋白質的一些溶劑不能溶解包涵體蛋白質時,可以使用溶解的方法把膜蛋白質除去。最經常的選擇性溶解膜蛋白質的方法是利用鰲合金屬的試劑如EDTA、中性的去垢劑如Triton X-100,或者一些鹽如,脫氧膽酸鹽,或者弱離液劑,如2mol/L的脲。Babbit等人發現經過前面的步驟,可以提高肌氨酸激酶的復性收率,活性提高的原因是使用弱的去垢劑辛基糖除去了蛋白質酶。
離心和高壓勻漿都有可能產生泡沫,所以必須在工業規模中避免這種無謂的損失。有些工業生產中,分離以前在發酵罐中把細胞殺死使細胞內的物質釋放出來,並同時溶解或者不溶解包涵體蛋白質。這種線上殺死細胞的方法己經用來生產兩種蛋白質:在鹼性條件下溶解類胰島素生長因子和高溫、酸性條件下生產重組的抗真菌肽段。這種方法的優點是可以節省操作時間以及能量消耗,僅僅需要一步離心的步驟。最經常使用的殺死細胞的試劑有苯乙醇,辛酸,氯仿,甲苯等。殺死細胞時也有缺點,溶解的目標蛋白質中含有蛋白質或者非蛋白質的物質,降低了蛋白質的純度;如果目標蛋白質沒有被溶解,則殺死細胞會使可溶蛋白質產生沉澱,使得包涵體的純化比較困難,或者破壞包涵體內部的重組蛋白質。
溶解
維持包涵體內蛋白質結構的作用力是分子內的作用力,這種作用力也維持天然蛋白質的穩定性的結構。先前有報導這種作用力是共價鍵結合的,但是,現在趨向於一致,就是維持包涵體內部的蛋白質的緊密的結構的是非共價鍵的作用力。二硫鍵,無論是正確的還是錯誤的二硫鍵,在維持內部蛋白質的緊密的結構中都沒有發揮直接的作用。最經常的獲得活性蛋白質的第一步是溶解這些包涵體蛋白質,溶解液是使這些包涵體蛋白質完全變性的成分,當蛋白質被溶解以後,則進入到蛋白質的體外摺疊的過程。
 包涵體溶解
包涵體溶解1. 遵循標準
包涵體蛋白質的溶解同樣是一個工藝的關鍵的步驟。溶劑的選擇會影響後續的操作、最終的各種蛋白質的收率以及最終的成本,必須遵循以下的標準:
(1)快速溶解的動力學;
(2)與蛋白質的結合是可逆的;
(3)對細胞碎片的分離方法沒有干擾作用;
(4)對溫度沒有依賴作用;
(5)抑制蛋白質酶的降解作用;
(6)與蛋白質的氨基沒有化學修飾作用;
(7)在可能的情況下,選擇最低的溶解濃度和廉價的溶劑,並適於以後的復性方法。
2. 溶解包涵體的試劑
最經常使用溶解包涵體的試劑包括離液劑或者去垢劑。
最經常使用的溶解和製備蛋白質的離子型的離液劑最早於1969年Hatefi等人發展的離子型的去垢劑如SDS是另外一種溶解包涵體蛋白質和膜蛋白質的試劑,但是一般不用來大規模的生產,而是用來定性。除了強酸、強鹼和利用有機溶劑來提取疏水性很強的蛋白質以外,其他的變性方法如非可逆的共價修飾在工業的大規模生產中很少用到。一旦蛋白質被溶解,蛋白質中的巰基很容易快速地氧化並形成共價的聚集體或者分子內錯配的二硫鍵,然後這些蛋白質就不能再進行摺疊。為了防止氧化,可以使這些基團或者利用緩衝液中含有低分子量的疏基試劑保持在還原的狀態或形成磺酸鹽或者形成混合的二硫鍵。
 蛋白質結構圖
蛋白質結構圖(1)去垢劑
去垢劑是一種最經濟的溶解包涵體蛋白質的方法,一個最大的優點是溶解的蛋白質有可能保持全部的生物活性,說明在此條件下保持了蛋白質的四級結構。最重要的是稀釋以後蛋白質的聚集比其它溶劑生成的很少。陽離子型、陰離子型的和非離子型的去垢劑都可以使用,使用時的濃度一般高於去垢劑的臨界膠束濃度(CMC ),通常是0.5-5%。
SDS僅僅在大量生產牛生長激素、干擾素和白介素-2中用到。SDS由於具有較低的臨界膠束濃度(CMC)而使得結合到蛋白質分子上的SDS比較難於除去。由於N-十二烷肌氨酸它的CMC比SDS高0.4%,也被用來溶解包涵體蛋白質並可用稀釋的方法使蛋白質復性,殘餘的去垢劑可以使用陰離子交換色譜或者超濾的方法除去。這種去垢劑是一種比較溫和的去垢劑,可以選擇性地溶解一些包涵體,但是不能溶解完全的變性的蛋白質的聚集體和大腸桿菌的內膜的蛋白質分子。使用去垢劑一個主要的缺點是對以後的純化和復性的步驟的干擾,去垢劑結合到蛋白質上的強度大離子交換色譜復性蛋白質小不同,比較難於除去,並干擾離子交換和疏水相互作用色譜的過程,在變性的濃度時超濾膜會吸附這些變性劑。所以復性後需要儘量洗滌這些去垢劑,也可以使用環狀糊精鏈狀糊精或者環狀澱粉從復性緩衝液中提取去垢劑。
一個不容忽視的問題是去垢劑可以溶解全部的膜蛋白質中的蛋白質酶,這些蛋白質酶的活性在去垢劑的存在的情況下被活化,可能造成溶解和復性過程的收率的降低。蛋白質復性的收率可以通過以下的方法來提高:
a)先期使用可以溶解膜蛋白質但是不溶解包涵體蛋白質的溶劑儘量洗滌包涵體蛋白質;
b)包涵體的含有的菌體碎片被完全除去;
c)溶解包涵體的液體中含有蛋白質酶的抑制劑,如EDTA,苯甲基磺醯氟(PMSF )等 。
(2)離液劑
其它的離液劑也被用來溶解包涵體蛋白質,最主要的溶解包涵體蛋白質的離液劑是鹽酸胍和尿素,這是最經常使用的溶解試劑,一般情況下選擇6-8mo1/L的濃度,蛋白質濃度在1-10mg/mL。
在溶解色氨酸合成酶A的過程,發現陽離子的溶解能力順序是Gdm+ > Li+ > K+ > Na+,陰離子的順序是SCN- > I- > Br- >Cr-。一些離液劑由於它們的溶液比鹽酸胍和尿素有更高的密度和黏度而不適合用於溶解包涵體,因為利用離心和色譜分離起來比較困難。
為了溶解包涵體蛋白質需要的尿素或者鹽酸胍的濃度根據蛋白質的不同而不同。如果蛋白質天然形態需要溶解的變性劑的濃度不能獲得,則在溶解包涵體時需要首先確定離液劑的濃度。
 大腸桿菌
大腸桿菌鹽酸胍由於比較貴,所以一般用來溶解一些附加值比較高的藥物蛋白質分子,選擇鹽酸胍作為溶解試劑,是因為鹽酸胍是一種比脲更為強烈的變性劑,甚至可以溶解脲所不能溶解的包涵體;尿素,由於可能被自發的形成的氰酸鹽或者已有的氰酸鹽的污染,特別是在鹼性環境中,從而造成蛋白質的自由的氨基被不可逆的修飾。消除此種影響的方法是用陰離子的緩衝系統如Tris-HCl溶解脲或者脲在使用之前利用陰離子交換色譜純化,並且配製的溶解和復性的緩衝液在當天使用。脲溶液中影響蛋白質變性的因素與鹽酸胍的不同。溶在脲中的蛋白質受到pH和離子強度的影響,從而影響電荷的蛋白質殘基之間的電荷作用,但是由於鹽酸胍含有高濃度的離子強度,所以這兩個因素的影響很少。
(3)混合溶劑
一般情況下去垢劑並不聯合使用,Lilly等人發現去垢劑和尿素的混合液有效的摩爾濃度較低。尿素和去垢劑型的鹽混合可以使蛋白質變性,但是尿素和非去垢劑的鹽如氯化鈉反而降低包涵體蛋白質的溶解性,所以要避免使用。
去垢劑結合其他的試劑或者溶解增強劑也被使用,發現尿素和乙酸,尿素和二甲亞楓,尿素和高pH等是比較有效的溶解包涵體蛋白質的方法。
高壓(1-2kbar)、超聲也可以溶解包涵體蛋白質,此時使用的溶解試劑濃度可以比較低,便於後續的復性步驟。
3. 極端pH
 細胞
細胞酸鹼度也是比較廉價的有效的溶解包涵體的方法。最經常使用酸的是有機酸,濃度在5-80%之間。Gavif和Better使用低的(pH≤2.6)和高溫(85℃ )溶解抗真菌的重組蛋白質的膚段,低溫和高PH需要溶解時間要長。Reddy和合作者也使用20%乙酸溶解一種麥芽糖結合的蛋白質。但是,同樣的一些不可逆的修飾作用或者酸降解會在極端pH下發生,所以此種方法並不是經常使用的溶解包涵體的方法。
高pH(≥12)也被用來溶解生長激素和原胰島素。在高pH下一些蛋白質同樣可能發生非可逆的變性,原因在於半胱氨酸在鹼性條件下的脫硫過程。所以這種方法儘管比較簡單、廉價,同樣僅僅用於一些特定的蛋白質,特別對於藥用蛋白質一般不採用這種方法。
包涵體與蛋白質
1. 真核細胞表達外源蛋白質的缺點
一些蛋白質需要翻譯後的修飾,如糖基化,則必須選用真核細胞。但是無論是正在臨床的還是市場上的蛋白質藥物,主要的是以大腸桿菌作為宿主細胞。
 核內包涵體
核內包涵體(1)真核細胞的天然蛋白質的表達和外源蛋白質的表達,表達量相當少,即使有的蛋白質表達量高時,容易出現蛋白質的無活性的現象,或者形成聚集體;而原核細胞表達的蛋白質量比真核細胞大很多;
(2)相對於大腸桿菌,人類對於真核細胞的基因的了解還是有限的,而對大腸桿菌的基因了解的相當透徹,並能進行熟練的基因重組等操作對大腸桿菌的基因進行改造;
(3)真核細胞生長到高密度需要更長的時間(7天左右),並且細胞的最終濃度低,所以需要較大的培養容器,而大腸桿菌可以在幾個h以內達到108cells/mL;
(4)動物細胞生長的培養液的造價較高,並且大部分使用血清作為生長液的添加劑,從而提高了產品的造價並且不利於以後的純化步驟;
 rna包涵體
rna包涵體(5)原核細胞的堅硬的細胞壁,可以使用簡單的攪拌的方法完成傳熱、傳質過程,而大部分的真核細胞需要溫和的培養方法及複雜的操作以達到其對培養基的要求 。
基於以上的原因,真核細胞尤其是哺乳動物細胞表達外源蛋白質大部分處在研究階段,大部分重組蛋白質使用的表達載體是大腸桿菌細胞。大腸桿菌表達的外源蛋白質量相當大,有時達到細胞內蛋白質總量的5-20%,甚至達到50%以上。但是,大腸桿菌表達的蛋白質,當含量相當大時,有時由於原核細胞缺少分泌到胞外的機制或者摺疊的機制,最後表達的蛋白質往往以無活性的包涵體的形式存在。
2. 包涵體表達蛋白質的優越性
為了研究基因的功能,可以把體內的基因轉移到其他表達宿主中,研究基因表達性狀以確定基因的功能。但是,外源基因的表達,特別是真核生物基因在大腸桿菌中的表達,由於宿主菌缺乏此種基因表達的調控機制,細胞內的化學環境與其天然環境差異,如氧化還原環境、胞內pH、自由水的濃度,以及外源基因的導入,使得表達目的蛋白質的細胞在非正常狀態下生長,表達的蛋白質多數以無活性的包涵體的形式存在。
在下游生產過程中特別是在醫藥生產中,以包涵體形式表達的蛋白質具有一些優越性。
(1) 異源表達的蛋白質很容易被宿主細胞內的蛋白質酶降解,如果重組蛋白質以包涵體形式表達,由於包埋了酶攻擊的位點,可以最大限度地抵抗蛋白質酶的攻擊;
(2) 包涵體表達的蛋白質沒有活性,細胞破碎和以後的純化步驟不用考慮蛋白質的失活問題;
(3) 包涵體形式表達的蛋白質與宿主細胞的其他蛋白質的分離比可溶蛋白質的分離方法簡便,造價低,使用簡單的離心或者過濾的手段就能使包涵體與宿主細胞的其他蛋白質成分進行有效的分離;
(4) 有些表達的外源蛋白質對細胞有毒性或者致死,大量表達時會導致細胞的死亡,最終的細胞數量和產物相當低;而包涵體形式的蛋白質由於喪失了生物活性從而可以高效大量地表達;
(5) 對於以包涵體形式表達的產物可以比較容易進行線上觀測定性,含有包涵體蛋白質的細胞有較大的折射率,不必像可溶的蛋白質需要細胞破碎後使用酶學或者電泳學的方法進行鑑定。
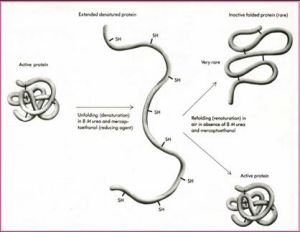 蛋白質復性
蛋白質復性包涵體的形成和蛋白質在等電點或者高鹽情況下的沉澱是不同的。在沉澱過程中,分子是通過分子表面的相互作用而形成的分子的聚集,無論在聚集的沉澱狀態還是在天然狀態,沒有明顯的構象的變化。因此,當溶液中的pH重新改變,或者沉澱重新溶解的時候,蛋白質的結構和活性會重新獲得。蛋白質的包涵體形式的聚集則不同於蛋白質的沉澱,把包涵體蛋白質直接溶解在天然的緩衝液中得不到天然的構象,無活性的聚集體與其天然活性形式之間沒有自然的平衡轉化的過程。包涵體的形成是由於范氏引力、疏水作用力和二硫鍵等作用力形成的,破壞這些作用力比較困難,溶解包涵體需要加入強的變性劑破壞多肽鏈之間的作用力,說明聚集體的多肽鏈之間的作用力遠遠大於普通的沉澱的分子間的作用力。當變性劑溶解的包涵體只是簡單地通過稀釋或者透析除去變性劑,而不是尋找合適的復性的條件的話,聚集體會重新形成,並且,不同於沉澱溶解的100%的活性的保留,包涵體的復性水平往往很低。所以,蛋白質沉澱是活性的天然蛋白質之間的作用力形成的,而包涵體的形成是在細胞內新合成的蛋白質肽鏈中間體而形成的,體外摺疊時的聚集體是摺疊過程中摺疊中間體形成的。這些摺疊的中間體並不一定是形成高級天然結構的中間體,從天然結構也不能推論出摺疊中間體的構象。
參考文獻
[1]於文國,陶秀娥. 包涵體蛋白復性及其影響因素[J]河北工業科技, 2007,(05).
[2]寧雲山,李妍,王小寧包含體蛋白質的復性研究進展.生物技術通訊,2001, 12( 3)23 7- 24 8
[3]王進,華子春,方勇,唐衛東,朱德煦,馮若,朱昌平,黃金蘭,陳兆華.功率超聲法對蛋白質包涵體的解聚.生物化學和生物物理進展,1995,22(6):543-545
[4]耿信篤.高效疏水色譜在蛋白質復性中套用.科學通報,1994,44(19):2046-2049
[5]萬雪, 王磊, 寧官保. 包涵體及其復性研究概況[J]. 畜牧獸醫科技信息, 2005,(02)
[6]井明艷, 孫建義. 蛋白質的摺疊調控與包涵體的形成[J]. 浙江大學學報(農業與生命科學版), 2004,(06)
[7]方敏,黃華樑. 包涵體蛋白體外復性的研究進展[J]生物工程學報, 2001,(06).
[8]馮小黎. 重組包涵體蛋白質的摺疊復性[J]生物化學與生物物理進展, 2001,(04).
[9]凌明聖,許祥裕,丁樹標. 以包涵體形式存在的重組蛋白的純化和體外摺疊[J]中國生化藥物雜誌, 1995,(03).
[10]楊曉儀,林鍵,吳文言. 重組蛋白包涵體的復性研究[J]生命科學研究, 2004,(02).
[11]羅惠霞,李敏,王玉炯. 包涵體蛋白復性的幾種方法[J]. 生物技術通報, 2007,(05).
[12]高飛,范清林,鄒文藝,宋禮華. 包涵體蛋白的層析復性技術研究進展[J]. 生物技術通報, 2006,(02).
[13]艾恆,莫書榮,魯波. 包涵體研究進展[J]. 中國醫學文摘.內科學, 2006,(02).
[14]鄒平. 重組包涵體蛋白質復性[J]. 廈門科技, 2005,(05).
[15]吉清,何鳳田. 包涵體復性的研究進展[J]. 國外醫學.臨床生物化學與檢驗學分冊, 2004,(06).
[16]高永貴,關怡新,姚善涇. 包涵體蛋白的變復性研究[J]. 科技通報, 2003,(01).
[17]Sunitha K, Chung B H, Jang K-H et al. Refolding and purification of zymomonas mobilis levansucrase produced as inclusion bodies in fed-batch culture of recombinant Escherichia coli .Protein Expr Purif. 2000, 18 :338-393 .
[18]DE BERNARDEZ E C. Protein refolding for industrial process .Current Opinion in Biotechnology. 2001, 12(2) :202 207 .
[19]http://baike.baidu.com/view/344949.html?wtp=tt
