原理
核酸分子雜交是基因診斷的最基本的方法之一。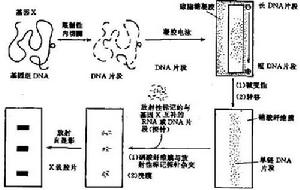 基因診斷技術
基因診斷技術一、基因探針
基因探針(probe)就是一段與目的基因或DNA互補的特異核苷酸序列,它可以包括整個基因,也可以僅僅是/基因的一部分;可以是DNA本身,也可以是由之轉錄而來的RNA。
1.探針的來源 DNA探針根據其來源有3種:一種來自基因組中有關的基因本身,稱為基因組探針(genomic probe);另一種是從相應的基因轉錄獲得了mRNA,再通過逆轉錄得到的探針,稱為cDNa 探針(cDNa probe)。與基因組探針不同的是,cDNA探針不含有內含子序列。此外,還可在體外人工合成鹼基數不多的與基因序列互補的DNA片段,稱為寡核苷酸探針。
2.探針的製備 進行分子突變需要大量的探針拷貝,後者一般是通過分子克隆(molecular cloning)獲得的。克隆是指用無性繁殖方法獲得同一個體、細胞或分子的大量複製品。當製備基因組DNA探針進,應先製備基因組文庫,即把基因組DNA打斷,或用限制性酶作不完全水解,得到許多大小不等的隨機片段,將這些片段體外重組到運載體(噬菌體、質粒等)中去,再將後者轉染適當的宿主細胞如大腸肝菌,這時在固體培養基上可以得到許多攜帶有不同DNA片段的克隆噬菌斑,通過原位雜交,從中可篩出含有目的基因片段的克隆,然後通過細胞擴增,製備出大量的探針。
為了製備cDNA 探針,首先需分離純化相應mRNA,這從含有大量mRNA的組織、細胞中比較容易做到,如從造血細胞中製備α或β珠蛋白mRNA。有了mRNA作模板後,在逆轉錄酶的作用下,就可以合成與之互補的DNA(即cDNA),cDNA與待測基因的編碼區有完全相同的鹼基順序,但內含子已在加工過程中切除。
寡核苷酸探針是人工合成的,與已知基因DNA互補的,長度可從十幾到幾十個核苷酸的片段。如僅知蛋白質的胺基酸順序量,也可以按胺基酸的密碼推導出核苷酸序列,並用化學方法合成。
3.探針的標記 為了確定探針是否與相應的基因組DNA雜交,有必要對探針加以標記,以便在結合部位獲得可識別的信號,通常採用放射性同位素32P標記探針的某種核苷酸α磷酸基。但近年來已發展了一些用非同位素如生物素、地高辛配體等作為標記物的方法。但都不及同位素敏感。非同位素標記的優點是保存時間較長,而且避免了同位素的污染。最常用的探針標記法是缺口平移法(nicktranslation)。
探針的標記也可以採用隨機引物法,即向變性的探針溶液加入6個核苷酸的隨機DNA小片段,作為引物,當後者與單鏈DNA互補結合後,按鹼基互補原則不斷在其3’OH端添加同位素標記的單核苷酸,這樣也可以獲得比放射性很高的DNA探針。
二、限制性核酸內切酶
限制性核酸內切酶(restrictionendonuclease),又簡稱限制酶或內切酶。它們是基因工程和基因診斷重要的一類工具酶。它們的發現和套用為從基因組中分離目的基因提供了必要的手段.限制酶能特異地識別和切割特異的核苷酸序列,將雙鏈DNA切成較小的片段。酶切後目的基因可能完整地或部分地保存於某一DNA片段上,並被分離出來。
限制酶主要來源於原核生物,是一組能水解DNA磷酸二酯鍵的酶。迄今已發現的限制酶多達數百種,分為三類。在基因工程中使用的主要是第二類。限制酶根據其來源命名。
每種限制酶識別和切割的通常為4-6個核苷酸序列,稱為限制性位點(restriction sites)或切點。限制酶切割雙鏈DNA的方式有兩種,產生的末端也有兩種:第一種是交錯切割,即兩條鏈的切點不在同一水平而是相隔數個鹼基,故斷口產生兩小段自身互補的單鏈,這種末端容易互補連線,稱為粘性末端(cohesive terminus);第二種為平整切割。
限制酶的上述特性在基因工程和基因診斷中具有重要用途:①首先不論DNA的來源如何,用同一種內切酶切割後產生的粘性末端很容易重新連線,因此很容易將人和細菌或人和質粒任何兩個DNA片段連線在一起,即重新組合,這是重組DNA技術的基礎。②人類的基因組很大,不切割無法分析其中的基因。限制酶能把基因組在特異的部位切開,即切割不是隨機的,因而從每個細胞的基因組得到的是相同的一組長度各異的片段。這些可能含有某一基因的片段可用電泳分離,並加以研究。③由於限制酶的特異性,如果識別位點的鹼基發生了改變,限制酶將不再能切割;同樣,鹼基的改變也可能導致出現新的酸切位點。在人類基因組中,這兩種情況是十分常見的,而切點的消失或出現將影響獲得的DNA片段的長度,表現為限制性片段長度多態性(RFLP),這在基因的連鎖診斷中具有極重要的意義。
三、限制性片段長度多態性
一個人的兩套單倍體DNA是不完全相同的,一般每100-500個鹼基對就有一個是不相同的。換言之,如果把兩套基因組DNA(各3.2×109bp)排列起來,那么平均有1000萬處不同,它們多位於內含子序列中。實際上,除單卵雙生子外,人群中沒有兩個個體的基因組DNA是完全相同的。
DNA的多態性雖可通過DNA測序檢出,但用限制酶消化卻是最常用的檢測方法。
1.RFLP由於鹼基的變異可能導致酶切點的消失或新的切點出現,從而引起不同個體在用同一限制酶切時,DNA片段長度出現差異,這種由於內切酶切點變化所導致的DNA片段長度的差異,稱為限制性片段長度多態性(restriction fragmentlength polymporphism, RFLP)。RFLP反映了常見的個體間DNA核苷酸的可遺傳性變異,它按照孟德爾方式遺傳。RFLP可用Southern印跡雜交法檢出。用Southern雜交檢出RFLP時,如探針跨越切點,則被切開的兩個片段均可與探針雜交,從而顯示兩條雜交帶。
2.兩點RFLP
(1)點多態(point polymorphism):是由於單個或少數鹼基的改變引起酶切點的出現或消失所致的RFLP。上述的RFLP即屬於這一類。它們屬經典的RFLP。在人類基因組中已發現數以百計的此類多態位點。
(2)數目變異的串連重複(variable number tandem repeats,VNTR):上述經典的單個鹼基取代所致的RFLP一般只能檢測到一種雜合性的兩種形式,即“有”或“無”某個限制酶切位點,而且每個位點在人群中的雜合子頻率通常不會超過50%,當被測個體為純合狀態時,利用RFLP就無法得到所需要的多態信息。此外,在整個基因組中,這類RFLP目前發現的數量還有限,並分布不勻。
但是,在人類基因組中還存在一類DNA重複序列,稱為小衛星DNA。它們分布十分廣泛,每一個單位通常只有16-28bp長,但其重複次數在人群中是高度變異的。當用限制酶切割VNTR區時,只要酶切點不在重複區內,就可能得到各種長度不同的片段與小衛星DNA不同,另一類重複序列是衛星DNA。它們的基本序列有1-6bp,如(TA)n、(CGG)n等,通常重複10-60次並呈高度的多態性。
VNTR具有高度的變異性,同時也是按照孟德爾方式遺傳的,因此是很好的遺傳標記,由於它們類型眾多和在基因組中分布廣泛,因而在基因連鎖診斷中套用日益廣泛。
基因診斷的對象
1、病原生物的侵入:一般侵入體內的病原生物可通過顯微鏡檢查各免疫學方法進行診斷。但是,直接檢測病原生物的遺傳物質可以大大提高診斷的敏感性。而肝在無法得到商業化抗體時,基因診斷就成為檢測病原微生物感染,尤其是病毒感染的唯一手段。此外,由於基因鹼基配對原理的基因診斷可直接檢測病原微生物的遺傳物質,所以診斷的特異性也大為提高。目前,基因診斷已在病毒性肝炎,愛滋病等傳染病的診斷中發揮了不可代替的作用。
2、先天遺傳性疾患:已有多種傳統的遺傳性疾患的發病原因被確定為特定基因的突變。例如:苯丙氨酸羥化酶基因突變可引起苯丙酮尿症:腺苷脫胺酶基因突變可引起重症聯合免疫缺陷症(SCID);而淋巴細胞表面分子CD40或其配體(CD40L)基因突變則可引起無丙種球蛋白血症。這類疾病的診斷除了仔細分析臨床症狀及生化檢查結果外,從病因角度作出診斷則需要用基因診斷的方法檢測其基因突變的發生。此外,有些病因尚不清楚的疾病,如高血壓、自身免疫性疾病等,都可能與某個或某些遺傳位點的持有或改變有並。用基因診斷的方法檢測這些位點的改變,不僅對臨床診斷,而且對疾病的病因和發病機理的研究都具有重要的意義
3、後天基因突變引起的疾病:這方面最典型的例子就是腫瘤。雖然腫瘤的發病機理尚未完全明了,但人們可以初步認為腫瘤的發生是由於個別細胞基因突變而引起的細胞無限增殖。無論是抑癌基因發生突變還是癌基因發生突變,如果確定這些改變的發生,都必須進行基因診斷。
4、其它:如DNA指紋、個體識別,親子關係識別,法醫物證等。
遺傳病
一、直接診斷和間接診斷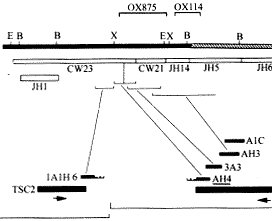 基因診斷
基因診斷二、基因異常與診斷方法的選用
各種遺傳病的基因異常是不同的,同一遺傳病也可以有不同的基因異常,但這些異常大體可分為基因缺失和突變兩大類型。後者包括單個鹼基置換、微小缺失或插入。近年來不發現一些遺傳病是由於基因內的三核苷酸重複順序增加引起的,根據對基因異常類型的了解,可以採用不同的診斷方法。如基因缺失可用基因探針雜交,PCR擴增直接檢測;點突變可用等位基因特異的ASO探針、SSCP等直接檢查。一般無需對家系成員進行分析。但條件是必需知道基因異常的性質,並肯定該異常與疾病之間的關係。然而,由於許多疾病的遺傳異質性,以及多數遺傳的基因異常尚屬未知,目前能直接診斷的病種雖日益增多,但仍然是比較有限的。
許多遺傳病的基因尚未分離克隆,或基因異常尚不清楚,因此還不能根據突變的性質進行診斷。但如果通過家系分析能證明某一DNA標記(無論是等位基因還是多態性位點或片段)與致病基因連鎖,則凡帶有該標記的成員都可能帶有致病基因,從而可作出間接的連鎖分析診斷。
套用連鎖分析診斷時應注意如下幾個問題:①基因與DNA標記之間可能發生重組。因此連鎖分析的準確性取決於DNA與致病基因連鎖的緊密程度,連鎖愈緊密,可靠性愈高,故應採用儘量靠近致病基因,即連鎖緊密的標記,或採用多個遺傳標記以儘可能排除重組。由於可能存在重組,連鎖分析不能完全確定致病基因是否存在,而是指出存在可能性或機率的大小。如果標記距基因有5cM,即重組率為5%,則作出診斷時尚有5%誤差的可能,即只有95%的把握作出肯定或否定的結論。②選用的遺傳標記在人群中的雜合度。如果標記的雜合度在人群中很低,即多數個體為純合子,則這種標記用處不大。因為如家系中關鍵成員不能提供致病基因在哪一條染色體上的信息,常使連鎖分析無法進行,因此需選用人群中雜合度高的標記。③在連鎖分析時,有時只分析一個多態位點還不能把某一家系中帶有致病基因的染色體與正常染色體區分開來。這通常是由於關鍵成員如待診者的父母是純合子,不能提供必要的信息,這時可同時分析更多的多態位點,即作單倍型(haplotype)分析。單倍型是指一條染色體上兩個或兩個以上多態位點的狀態的組合。兩條染色體多個位點上都純合的機率畢竟是很小的,因此單倍型有助於區分兩條常染色體,並追蹤致病基因的分離情況。
三、遺傳病的基因診斷舉例1.基因缺失型遺傳的診斷
(1)α地貧的基因診斷:α地貧主要是由於基因缺失引起的,缺失的基因可以由1-4個。正常基因組用BamHⅠ切割,可以得到一個14kb的片段,而缺失一個α基因時切點向5’端移位,得到一條10kb的片段。因此,當用α基因探針與基因組DNA進行Southern雜交時(圖13-8),在α地貧2可見一條14kb和一條10kb的帶,在正常人可見一條雙份的14kb的帶,而在α地貧1則見一條單拷貝的14kb帶,血紅蛋白H病時只有一條10kb的帶的,而在Barts水腫胎時,則無任何雜交帶。
一種較簡便的方法是直接用α探針進行斑點雜交,自顯影后根據斑點深淺的不同也可以對α地貧作出診斷。更為簡單的方法是PCR診斷,即在α基因缺失範圍內設計一對引物,然後PCR擴增胎兒的DNA,如為Barts 水腫胎,則無擴增產物,電泳後無任何帶紋,從而可建議進行人工流產,但此法不能診斷其它類型的地貧(除非另設計引物用作PCR)。
(2)DMD/BMD的缺失型診斷:DMD/BMD是一種Ⅹ連鎖隱性遺傳的神經肌肉系統受累的致死性遺傳病(參閱第四章)。DMD/BMD有70%左右為缺失型。此基因很大,缺失可發生在不同部位,因此應儘可能採用多對引物作PCR擴增(多重PCR)來檢測。如擴增產物電泳後發現有帶紋的缺失,即可作出診斷並對缺失定位(圖13-9),在進行產前診斷時,一般可先通過檢測家系中有關成員,即確定先證者的缺失區,然後有針對性地作PCR擴增,包括缺失部分的兩端,以判斷胎兒或有關患兒是否也獲得了相同的基因缺失,但非缺失型不能用此法查出。
2.點突變型遺傳病的基因診斷2
(1)鐮形細胞性貧血的基因診斷:已知突變基因是編碼β珠蛋白鏈的第6位密碼子由GAG變為GTG,從而使纈氨酸取代了甘氨酸,因此可用如下方法進行診斷。
1)RFLP診斷:已知限制酶MstⅡ切割的識別順序是CCTNAGG,它能切割正常β鏈中CCTGAGG序列,但不能切割突變了的CCTGTGG(A→T)。這樣,由於突變消除了一個切點,使內切酶長度片段發生了改變,通過電泳,就可以區別正常的βA和βS。
2)ASO探針診斷:由於突變部位和性質已完全明了,也可以合成寡核苷酸探針,用32P標化來進行診斷。此時需要合成兩種探針,一種與正常βA基因序列完全一致,能與之穩定地雜交;另一種與突變基因序列一致,能與βS基因穩定雜交,但不能與正常的βA基因雜交。根據雜交結果,就可以把發生了突變的βS基因檢測出來。
PCR技術問世以來,ASO診斷又有新的改進,即先PCR擴增長約110bp的基因片段,然後再與ASO探針雜交。這樣可減少目的基因DNA用量,並降低與基因組DNA雜交時的非特異性信號。
3.基因異常不明的遺傳病的診斷
成年型多囊腎病(adult polycystic kidney disease,APKD)是一種常染色體顯性遺傳病,發病率高,約1000人中有1名致病基因的攜帶者,起病較晚,多在30歲以後,主要為腎和肝中出現多發性囊腫,臨床表現為腰疼、蛋白尿、血尿、高血壓、腎盂腎炎、腎結石等,最終可導致腎功能衰竭和尿毒症。本病基因定位在16p13,與α珠蛋白基因3’端相鄰,但致病基因尚未克隆,基因產物的生化性質和疾病發病機理也尚未闡明。因此,只能用連鎖分析來進行基因的發病前診斷和產前診斷。由於通過家系分析,已證實APKD的致病基因與α珠蛋白基因3’端附近的一段小衛星DNA序列即3’HVR(3’ hypervariable region)緊密連鎖,而後者在人群中具有高度多態性,因此可以通過RFLP連鎖分析進行診斷。
常用技術
當細胞的基因組DNA用特定的內切酶如Eco RⅠ切割時,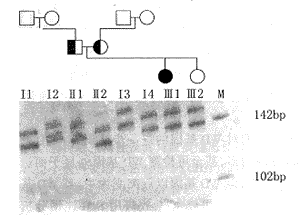 基因診斷
基因診斷然而許多長短不同的DNA片段混合在一起是很難分析的。因此首先必需將它們按大小(長短)分離開來,這可藉助凝膠電泳來完成。在電泳時,分子量愈小的片段的遷移愈快,愈大的片段愈慢。因此,在電泳結束時可以獲得一個由大到小連續的帶譜(smear),而由許多細胞基因組得來的某一特定片段,因其長度相同將處於同一位置,有利於檢出。但凝膠易碎且操作不便。英國科學家Southern首創印跡法克服了上述困難。
一、Southern印跡法(Southernblot)
基本原理是:硝酸纖維膜或尼龍濾膜對單鏈DNA的吸附能力很強,當電泳後凝膠經過DNA變性處理,覆以上述濾膜,再於其上方壓上多層乾燥的吸水紙,藉助它對深鹽溶液的上吸作用,凝膠上的單鏈DNA將轉移到濾膜上。轉移是原位的,即DNA片段的位置保持不變。轉移結束後,經過80℃烘烤的DNA,將原位地固定於膜上。
當含有特定基因片段已原位轉移到膜上後,即可與同位素標記了的探針進行雜交,並將雜交的信號顯示出來。雜交通常在塑膠袋中進行,袋內放置上述雜交濾膜,加入含有變性後探針的雜交溶液後,在一定溫度下讓單鏈探針DNA與固定於膜上的單鏈基因DNA分子按鹼基到互補原理充分結合。結合是特異的,例如只有β珠蛋白基因DNA才能結合上β珠蛋白的探針。雜交後,洗去膜上的未組合的探針,將Ⅹ線膠片覆於膜上,在暗盒中日光進行放射自顯影。結合了同位素標記探針的DNA片段所在部位將顯示黑色的雜交帶,基因的缺失或突變則可能導致帶的缺失或位置改變。
分子雜交是基因探測的基礎,除了用印跡雜交外,還有斑點雜交法。即將DNA樣品變性後直接點在硝酸纖維濾膜上,再與探針雜交,或者將細胞或病毒點在膜上,菌落或菌斑原位地吸附在膜上,經過變性處理,再進行雜交。斑點雜交多用於病原體基因,如微生物的基因,但也可用於檢查人類基因組中的DNA序列。
二、聚合酶鏈反應
近年來,基因分析和基因工程技術有了革命性的突破,這主要歸功於聚合酶鏈反應(polymerase chain reaction,PCR)的發展和套用。套用PCR技術可以使特定的基因或DNA片段在短短的2-3小時內體外擴增數十萬至百萬倍。擴增的片段可以直接通過電泳觀察,也可用於進一步的分析。這樣,少量的單拷貝基因不需通過同位素提高其敏感性來觀察,而通過擴增至百萬倍後直接觀察到,而且原先需要一、二周才能作出的診斷可以縮短至數小時。
首先應按照欲檢測的DNA的5’和3’端的鹼基順序各合成一段長約17-20餘個鹼基的寡核苷酸作為引物(primer),其次是將待檢測的DNA變性後,加入四種單核苷酸(dNTP)、引物和耐熱聚合酶。在較低的溫度,引物將與待擴增的DNA鏈復性結合,然後的聚合酶的作用下,利用溶液中的核苷酸原料,不斷延伸合成新互補鏈,這樣,一條DNA雙鏈就變成了兩條雙鏈。若繼續按照變性(92-95℃)→復性(40-60℃)→引物延伸(65-72℃)的順序循環20至40個周期,就可以得到大量的DNA片段。理論上循環20周期可使DNA擴增2n,即100餘萬倍。PCR反應特異性強,靈敏度高,極微量的DNA即可作為擴增的模板得到大量的擴增片段。毛髮、血痕,甚至單個細胞的DNA即可供PCR擴增之用。因此它用於病原體DNA的檢查、腫瘤殘留細胞的檢出、罪犯或個體遺傳物質的鑑定以及遺傳病的基因診斷等。
已可對一系列的遺傳病進行PCR診斷。如果疾病是由基因缺失引起的(如α地貧),則在缺失兩端設計一對引物進行擴增,就不會得到擴增產物或只能得到縮短了的擴增產物。如果疾病是由點突變引起的,而突變的位置和性質已知,則在設計引物時使之包括突變部位,由於突變後的鹼基不配對,結果無擴增片段;或者在引物設計時於其3’端設計一個錯誤的核苷酸,使之與突變了的核苷酸配對,其結果是正常引物不能擴增,而用錯誤的引物能擴增,從而可對突變的存在作出判斷。
PCR技術目前有許多新的發展,用途日益擴大。例如,可用RNA為模板經過逆轉錄再行擴增的RT-PCR;改變兩引物濃度,使其相差100倍,結果得到大量單鏈產物,稱為不對稱PCR,其單鏈產物可用於序列分析;在一個反應中加入多對引物同時檢測多個部位的多重PCR等等。
三、擴增片段長度多態性
小衛星DNA和微衛星DNA的長度多態性可以通過PCR擴增後電泳來檢出,並用於致病基因的連鎖分析,這種診斷方法稱為擴增片段長度多態性(amplified fragment length polymorphism,Amp-FLP)連鎖分析法。PCR擴增後,產物即等位片段之間的差別有時只有幾個核苷酸,故需用聚丙烯醯胺凝膠電泳分離鑑定。此法多用於突變性質不明的連鎖分析。
四、等位基因的特異寡核苷酸探針診斷法
當基因的突變部位和性質已完全明了時,可以合成等基因特異的寡核苷酸探針(allele-specific oligonucleotide,ASO)用同位素或非同位素標記進行診斷。探針通常為長20bp左右的核苷酸。用於探測點突變時一般需要合成兩種探針,一種與正常基因序列完全一致,能與之穩定地雜交,但不能與突變基因序列雜交;另一種與突變基因序列一致,能與突變基因序列穩定雜交,但不能與正常基因序列穩定雜交,這樣,就可以把只有一個鹼基發生了突變的基因區別開來。
PCR可結合ASO,即PCR-ASO技術,即先將含有突變點的基因有關片段進行體外擴增,然後再與ASO探針作點雜交,這樣大大簡化了方法,節約了時間,而且只要極少量的基因組DNA就可進行。
五、單鏈構象多態性診斷法
單鏈構象多態性(signlestrand conformation polymorphism,SSCP)是指單鏈DNA由於鹼基序列的不同可引起構象差異,這種差異將造成相同或相近長度的單鏈DNA電泳遷移率不同,從而可用於DNA中單個鹼基的替代、微小的缺失或手稿的檢測。用SSCP法檢查基因突變時,通常在疑有突變的DNA片段附近設計一對引物進行PCR擴增,然後將擴增物用甲醯胺等變性,並在聚丙烯醯胺凝膠中電泳,突變所引起的DNA構象差異將表現為電泳帶位置的差異,從而可據之作出診斷。
PCR-SSCP法具有能快速、靈敏地檢測有無點突變或多態性的優點,但如欲闡明突變的鹼基性質,則需作序列分析。
