概述
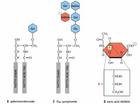 半乳糖腦苷類脂
半乳糖腦苷類脂克拉伯病(Krabbedisease)於1916年由丹麥兒科醫師Krabbe首先報導因此稱為Krabbe病,依據其臨床特點,亦稱為嬰兒家族性瀰漫性硬化(infantilefamilialdiffusesclerosis),為常染色體隱性遺傳代謝性疾病突變基因位於14p。
 克拉伯病
克拉伯病克拉伯病的基因缺陷引起半乳糖腦苷-β-半乳糖苷酶缺乏,是導致主要累及腦白質的遺傳代謝性疾病。本病預後極差嬰兒型者常於1歲之內病故。晚發者可生存至10歲左右目前尚未查到權威性的較全面的發病率統計學資料。本病十分罕見有文獻報導發生率約為1/5萬新生兒。隨著病情發展臨床症狀體徵多樣特別是應注意合併的智慧型發育遲緩、失明、失聰、肺部感染、跌傷等。
流行病學
 克拉伯病
克拉伯病病因
 克拉伯病
克拉伯病克拉伯病是由於溶酶體中的半乳糖腦苷脂酶缺乏所致。該酶的編碼基因位於14q2l~q3l,其突變造成的酶活力缺陷使半乳糖腦苷脂不能降解成神經醯胺和半乳糖。其病理變化的特徵是:腦白質中有大量球形多核細胞(球形細胞是胞內充滿未降解的半乳糖腦苷脂的血源性巨噬細胞),髓鞘質和少突神經膠質大量喪失;腦白質中星形細胞神經膠質增生,外周神經通常可見到節段性脫髓鞘變、軸突退行性變、纖維化和巨噬細胞浸潤等病變。
臨床表現
 克拉伯病
克拉伯病嬰兒型患兒多數在3~6個月時發病,首發症狀以易激若、陣哭、淡漠或嘔吐等餵養困難為主,多數伴有營養不良;病情發展迅速,很快即出現進行性軀幹和四肢肌張力降低、肌陣攣、腱反射亢進和錐體索征陽性;1歲時即可發生眼震顫、斜視等,鏇即視神經萎縮、失明;少數患兒聽力亦喪失。晚期患兒肌張力亢進,常呈角弓反張,多數在2歲以內死於呼吸困難或肺部感染。
晚髮型約占全部krabbe病患者的10%~15%。發病年齡自15個月~10歲不等,但多數在5歲以前;臨床症狀與嬰兒型相似。初起出現進行性的行走困難,或痙攣性單側下肢癱瘓或偏癱最先出現,數周或數月後出現雙側錐體索征;半數以上患兒可見深腱反射消失、神經傳導速率減低等外周神經被侵犯的徵象。隨著病程延長,神經系統症狀日益加重,由於視神經脫髓鞘變所致的失明常見,患兒或遲或早出現智慧型衰退和行為異常,但癲癇發作不多見。病程長短不一,多數在起病2~5年後出現四癱和痴呆,少數可長達10~20年。
本病的實驗室特徵是:腦脊液中蛋白質含量異常增高;神經傳導速率降低;腦部磁共振掃描可見腦室周圍和頂枕葉部有脫髓鞘病變。確診必須依據對半乳糖腦苷脂酶活力的檢測,通常採用外周血白細胞進行;產前診斷可採取培養羊水細胞或絨毛活檢進行酶學檢測。
發病機制
 半乳糖苷酶
半乳糖苷酶診斷
 血白細胞檢查
血白細胞檢查檢查
 腦脊液常規檢查
腦脊液常規檢查實驗室檢查
2.血清培養的成纖維細胞中測定半乳糖腦苷β半乳糖苷酶活性缺乏。
其它輔助檢查
1.腦電圖呈非特異性慢波或局灶性慢波。
2.腦CTMRI掃描可見雙側內囊和基底核對稱性密度增高。
3.肌電圖檢查表現為去神經支配和運動感覺神經傳導速度減慢。
治療
迄今仍無滿意的治療方法,目前骨髓移植在Krabbe病鼠模型中已取得成功。本病確診是根據白細胞或皮膚成纖維細胞的酶活性測定。雜合子的酶活性在正常與患者之間。產前診斷已有可能。遺傳諮詢很重要。治療無特異方法,主要是支持療法和對症。溶酶體酶代替療法和骨髓移植仍在動物實驗階段。
預防
本病預後極差。嬰兒型者常於1歲之內病故晚發者可生存至10歲左右遺傳代謝性疾病治療困難,療效不滿意預防顯得更為重要預防措施包括避免近親結婚,推行遺傳諮詢、攜帶者基因檢測及產前診斷和選擇性人工流產等防止患兒出生。
