
病因
 亨丁頓舞蹈症
亨丁頓舞蹈症亨丁頓舞蹈症會造成亨丁頓舞蹈症的原因是腦細胞的神經原持續退化,這種退化會造成不能控制的運動,失去智慧型及情緒上的困擾,特別是影響到控制協調動作神經節時。在神經節內,亨丁頓舞蹈症會針對腦皮層神經原,特別是尾狀核特定攻擊。也會影響到控制思想,領悟力及記憶的腦部外層表面。
臨床表現
 神經
神經1.神經症狀
舞蹈樣症狀一般出現在智慧型障礙前。早期常為不規則的肌肉抽動,表現手指屈伸運動、點頭、面肌抽動呈怪相。進一步發展為面、頸、肢體和軀幹出現突然、無目的、強烈的不自主舞蹈樣動作。其特點是快速無規律,突然的有時像手足徐動症一樣緩慢而有節奏的運動。伴有發音不清與步態改變,也可以出現錐體外系症狀,患者常用同方向的隨意運動來偽飾。它屬於一種肌張力減弱、運動功能增強的錐體外系綜合徵。異常運動日益增劇導致明顯的扭轉樣動作與共濟失調。舞蹈症狀出現後智慧型障礙往往加重。
2.精神症狀
以情緒障礙為突出表現,尤以抑鬱常見。情感淡漠、遲鈍、抑鬱,常有自殺行為或人格改變,脾氣壞,喜歡爭吵,注意力渙散等。部分患者可出現偏執型精神分裂樣臨床徵象和精神分裂樣症狀,被害妄想,常有誇大和宗教色彩和人格改變,如易激惹、衝動、狂怒、行為怪癖等。亨廷頓病性痴呆常有抑鬱症狀,並有明顯自殺傾向。
智力障礙有時可作為首發症狀。早期患者注意力分散頗突出,可表現遲鈍、被動、淡漠、懶散,理解力差,工作效率下降或不能勝任工作等。以後出現痴呆症狀,發展緩慢。早期生活能力下降,有一定自知力,自述思考遲緩、健忘,記憶障礙較AD輕,判斷力往往受損,定向力相對保持。言語困難,失用、失認少見。晚期出現運動不能性緘默和呆傻,常緩慢進展。患者的記憶比其他認知功能受到影響較少,自知力常保持完好。病人在一段時期內能夠察覺自己智力的改變,主訴感到遲緩、健忘、腦子糊塗,被視為"皮質下痴呆"。晚期可出現明顯運動不能性緘默症。
檢查
 亨廷頓舞蹈症病人
亨廷頓舞蹈症病人1.簡易智力狀況檢查(MMSE)
由Folstein於1975年編制,評定計分標準,如回答或操作正確記“1”,錯誤記“5”,拒絕回答或說不會記“9”或“7”。主要統計“1”的項目總和(MMSE總分),範圍為0~300國際標準24分為分界值,18~24為輕度痴呆,16~17為中度痴呆,≤15分為重度痴呆。我國發現因教育程度不同臨界值也不同;文盲為17分,國小(教育年限≤6年)為20分,中學及以上為24分。
2.長谷川痴呆量表(HDS)
由長谷川和夫1974年制訂。共11個項目,包括定向力(2項)、記憶力(4項)、常識(2項)、計算(1項)、銘記命名回憶(2項)。該量表採用正向記分法,滿分為32.5分。原作者的臨界值定為:痴呆≤10.5分,可疑痴呆10.5~21.5分、邊緣狀態22.0~30.5分、正常≥31.0分,亦可按教育程度劃分正常值:文盲≤16分、國小<20分、中學以上<24分。
3.日常生活能力量表(ADL)
1969年Lawton和Brody制訂,主要用於評定受試者日常生活能力。ADL共分14項,評分為4級:①自己完全可以做;②有些困難;③需要幫助;④根本不能做。64分為滿分,總分≤16分完全正常,>16分有不同程度功能下降。單項分1分為正常,2~4分功能減退,有2項或2項以上≥3或總分≥22為臨界值,提示功能有明顯減退。我國常規總分18.5±5.5。
4.其他輔助檢查
頭顱CT和MRI可顯示尾核萎縮。正電子發射斷層掃描(PET)顯示尾核代謝明顯低下。
診斷
1993年,科學家發現基因檢查的方法來診斷亨丁頓舞蹈症。使用血液來分析DNA重複數的變異,重複數28以下沒有亨丁頓舞蹈症,而40以上有亨丁頓舞蹈症,在其間則為不確定的範圍。
亨丁頓舞蹈症患者眼睛無法協調或固定移動目標,眼睛不正常的移動也會因人而異。醫生也會要求作計算機斷層,掃描可提供腦部結構的微小變化,有亨丁頓舞蹈症時會顯示腦部的萎縮,特別是在尾狀核,並且有腦內的腦室空洞變大發生。
其他的檢查像是MRI(核磁共振)及PET,都是診斷亨丁頓舞蹈症的重要工具。亨丁頓舞蹈症檢查需要少量的血液檢查重複數,下一代可採取胎兒的組織來檢查,未來或可使用試管嬰兒篩選出正常的胚胎植入子宮。
發病
每人發病的時間都不同。成年或典型的亨丁頓舞蹈症會在中期發病,而青年期的亨丁頓舞蹈症會在20歲前,而目前已知CAG重複數最高為100者在2歲死亡。有些人在55歲以後發病,而這類病人常會與其他健康疾病所混淆,這些人會有憂鬱的情況,但仍然可能有敏銳的智慧型功能,像記憶及理解力。發病機制
雖然HD中腦部退化的機制尚未完全清楚,然而,腦細胞死亡的最終過程似乎是由一組胺基酸(我們稱之為興奮性胺基酸)所調節,這組胺基酸為由過度刺激造成精疲力竭的神經元所釋出,這些神經元最終會趨向死亡,此現象稱之為“興奮毒性細胞死亡”(excitotoxiccelldeath)。有研究將這些可活化興奮毒性接受器的化學物質注入動物體內會產生類似HD之選擇性神經元流失現象,這些動物的生化及行為改變亦極類似於HD,這些研究結果顯示:興奮毒性細胞死亡在HD中可能扮演極重要的角色。
HD患者紋狀體內中型刺狀神經元之能量代謝會受到影響,此不正常能量代謝狀況會快速地耗損神經元,使其易受興奮毒性的影響。粒腺體(mitochondria)為細胞的力量來源,其可經由一連串的化學變化(氧化磷酸化作用)而產生能量;氧化磷酸化的抑制劑如:3-nitropropionic acid(3NP)可造成類似HD之臨床症狀並提升乳酸濃度,由於3NP明顯可造成中型刺狀神經元之損傷,最後損及皮質神經元,故能量代謝異常亦可能與HD致病機制有關。
遺傳
 亨廷頓舞蹈症染色體
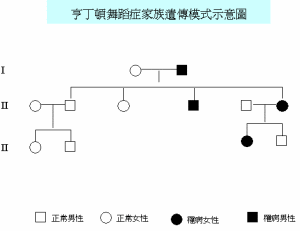
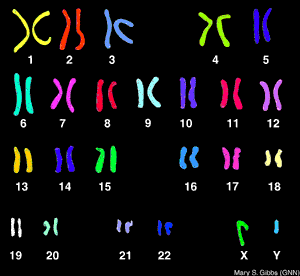
亨廷頓舞蹈症染色體人體內共有23對染色體,造成亨丁頓舞蹈症的基因是在第四對染色體,與性別無關,因此男人與女人得到的機會一樣。第四對染色體內的DNA基質重複排序數目太多時會造成亨丁頓舞蹈症,而亨丁頓舞蹈症會重複複製CAG為正常的10多倍。每個父母只提供半對染色體給下一代,因此下一代遺傳的機會為50%,假如下一代沒有遺傳到亨丁頓舞蹈症,就不會發病也不會遺傳到下下代,如果有亨丁頓舞蹈症則遲早會發病。而有些家庭有些小孩會得到亨丁頓舞蹈症,而有些不會,有一個小孩有亨丁頓舞蹈症並不代表另一個也會得到。而有些人家族史內並無亨丁頓舞蹈症但發病,原因可能為基因突變。
治療方法
藥物可幫助減輕情緒或動作的問題。但重要的是,藥物可以使症狀控制,但無法改變退化的發生,沒有任何治療可以停止或恢復亨丁頓舞蹈症。
精神藥物如Haloperidol可以緩和舞蹈動作或減少幻覺及突爆情緒,但如果亨丁頓舞蹈症有語言失調時,此藥物就不被使用,因為會使情況惡化如僵硬及緊繃。這類的治療藥物會有嚴重的副作用包含鎮定,所以要使用最低限量。大部分治療亨丁頓舞蹈症的藥品都有副作用如疲憊,無法休息,或過度興奮。
相關研究
1、美發現可延緩“亨廷頓舞蹈病”發作的膽汁酸美國研究人員於2002年找到了一條治療“亨廷頓舞蹈病”的新線索。他們在實驗鼠身上發現,一種膽汁酸能有效減輕這一疾病對腦神經細胞的損傷。明尼蘇達大學基恩等人在美國《全國科學院學報》上介紹說,在實驗中,他們選擇了一組攜帶亨廷頓舞蹈病致病基因,且呈現發病症狀的病鼠和一組健康鼠進行對比測試。
研究人員先給一半病鼠注射了牛磺熊脫氧膽酸(TUDCA),然後再讓注射了這種膽汁酸的病鼠、未接受注射的病鼠以及正常的實驗鼠,分別參加了走迷宮測試。結果顯示,接受膽汁酸注射治療的病鼠比未接受注射病鼠的能力強一半,且與健康鼠幾乎沒有區別。大腦檢查還發現,注射了膽汁酸的病鼠腦神經細胞死亡數比未接受注射的病鼠明顯要少。
2)美科學家發現剛果紅可治療亨廷頓舞蹈病
美國哈佛醫學院的科學家在2003年1月23日出版的《自然》雜誌上報告說,一種常用於醫學實驗的染料剛果紅,能夠阻止腦部一些異常蛋白質積聚,防治“亨廷頓氏舞蹈病”。剛果紅是醫學實驗中的一種常見的細胞標記染料。研究人員對亨廷頓氏舞蹈病患者腦部切片進行檢查時,通常用剛果紅對之進行染色。剛果紅能附著在異常蛋白質上,清晰地顯示出蛋白質塊的積聚區。哈佛醫學院科學家進行的腦細胞組織培養實驗表明,剛果紅染料不僅能附著在異常蛋白質塊上,還能減緩蛋白質的積聚過程,用剛果紅處理過的腦細胞不易死亡。帶有亨廷頓氏舞蹈病基因的實驗鼠,接受剛果紅染料注射後,也較少產生神經功能障礙。
這一發現為治療亨廷頓氏舞蹈病提供了新思路,但還需進一步研究改進才能將這種方法用於臨床。剛果紅還被用於標記阿爾茨海默氏症、克雅氏症等腦病樣本中的異常蛋白,因此它也可能有助於防治這些疾病。
3、與亨廷頓舞蹈病治療有關的關鍵蛋白
亨廷頓舞蹈病是由一個變異型亨廷頓基因(huntington gene)引起的,該基因可具有許多個胺基酸(稱作谷氨醯胺)拷貝的蛋白質。正常的亨廷頓蛋白含有10-25段谷氨醯胺序列。但如果有36段以上的谷氨醯胺序列時,該蛋白質的形狀即發生改變,並在神經元內形成大的叢群。這將殺滅大腦紋狀體部位的這些細胞,從而導致特有的協調力喪失和痴呆症 。
研究人員認為,如同亨廷頓蛋白一樣,CBP也包含一段較短的聚谷氨醯胺序列。這些序列相互吸引,使亨廷頓蛋白抓住CBP。為證實這一點,該研究小組創建了一種可剪斷的聚谷氨醯胺片段形式的CBP。當研究小組創建了一種可剪斷的聚谷氨醯胺片段形式的CBP。
Ross懷疑亨廷頓蛋白群“綁架”其他蛋白有可能也對其他大腦退化性疾病(如帕金森病)起作用。 美國的研究人員認為,治療亨廷頓舞蹈病的分子線索的發現有可能使開發保護大腦的藥物成為現實。倫敦Guy、King和聖托巴斯醫學院的神經遺傳學家Gillian Bates認為,這的確是一項重要的研究結果。
4、亨廷頓舞蹈病可能不完全取決於遺傳因素
2004年4月,墨西哥國立自治大學細胞生理學研究所馬謝烏•特里戈等三名科學家對亨廷頓氏病的起因進行研究並發表了相關文章。文章報導,亨廷頓氏病可能不完全取決於遺傳因素,有可能也與大腦中的毒蛋白有關。
傳統理論認為亨廷頓氏病是一種強遺傳因素引起的大腦神經退行性疾病,但它造成大腦認知功能衰退的病理機制至今還沒有完全被人們所認識。特里戈領導的科研小組對感染了亨廷頓氏病的小鼠模型進行實驗。他們給小鼠餵食一種對線粒體產生損傷的黴素,發現有少量谷氨酸鹽積聚在亨廷頓蛋白質上,使腦神經細胞內毒素水平不斷增加,導致腦神經細胞功能不全和數量減少,直至神經細胞全部死亡。 科研小組對該病患者腦部切片進行檢查時還發現,亨廷頓蛋白質里含有摺疊的核苷酸,會使大腦溝回區神經細胞的重量至少損失30%。因此他們認為,造成這種異常的“罪魁禍首”很可能是毒蛋白。
5、“包涵體”研究的新成果
當患者發病後,體內產生變異的亨廷頓蛋白質。該蛋白質在細胞內逐漸聚集,形成大的分子團,這些不溶水的分子團被稱為“包涵體”。業內主流認為,包涵體破壞了神經元,是亨廷頓舞蹈症的生化指標之一,因為患者腦細胞中有許多包涵體,而健康人腦細胞中卻沒有。而有的科研人員則認為包涵體是機體進行自我保護的一種機制,美籍華裔助理教授李沉簡是這一觀點的堅實支持者。他利用基因bank技術和乳糖操縱子技術,以及對小鼠的大腦切片的螢光顯微鏡觀察等多項試驗,證明了在他的實驗中,包涵體是機體的一種自我保護機制。 為了探明包涵體這種特殊物質的真正作用,美國加利福尼亞大學 的科學家發明了一種方法,觀察患有亨廷頓舞蹈病的實驗鼠的神經元形成、發育和消亡。
不過,實驗鼠的神經元是目前反映亨廷頓舞蹈病細胞活動的最佳模型。 此項實驗不但改變了科學家對包涵體的認識,可能也會改變亨廷頓舞蹈病的治療方法。很多醫藥廠家都在研製阻止變異亨廷丁蛋白質聚集成包涵體的藥物。由此項最新的研究推斷,這些藥不僅不會遏制病情,反而會加速該病的發展。醫生迄今尚未發現治療亨廷頓舞蹈病的有效手段。隨著病情的不斷加重,患者可能在10至25年內死亡。
6、細胞死亡
2005年9月,著名的神經醫學雜誌《Neuron》上刊登了一篇美國洛杉磯加大神經科學研究所助理教授楊向東的文章,他研究發現,從小鼠實驗發現腦細胞間互相影響會造成腦神經死亡,以致發生不能控制手足的亨廷頓舞蹈症疾病。他認為該病是因為腦細胞的死亡而造成的,假如能了解腦細胞死亡的原因,就可找到治療的方法。
他設計了將變異的人腦基因植入小鼠大腦的動物模型 ,並得到了嚴重亨廷頓舞蹈症狀的小鼠,從而在一定條件下證實了自己的觀點。
