流行病學
 瘋牛病
瘋牛病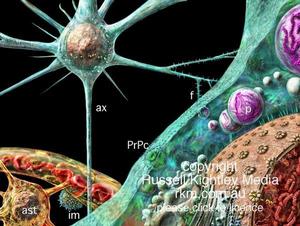 朊毒體病
朊毒體病傳染途徑
1.人對人傳染 如Kuru病的傳遞。但一般接觸傳染髮病尚無證據,而CJD病人對病人的醫源性傳染已有報告角膜移植、針電極腦電圖、外科手術傳染髮病均有報告。已有4例由於接受人類垂體生長激素代替治療發生CJD的報告。
2.動物傳染給人類利比亞人食用羊腦而使其發病率高,即羊瘙癢症傳給人類;瘋牛病(mad cow disease,MCD)的傳遞可能性雖然存在,但有待證實。
3.體記憶體在潛伏感染致病因子在外界環境因素誘發或遺傳因素作用下發病等尚未定論。
病因
非常規慢病毒致病因子被認為是一種澱粉樣蛋白原纖維(SAF)構成此種原纖維特殊蛋白顆粒被稱為朊病毒(PrP27-30)。這種慢病毒致病因子的性質既有病毒性傳染髮病特點,又有與常規病毒不同的理化特性和生物學特性。
理化特性表現為:①具有高度抗各種消毒滅活能力;②有抗高熱80~l00℃滅菌能力;③具有抗紫外線和其他各種射線作用能力;④常規電鏡看不到病毒,只有用特殊理化方法可發現原纖維和蛋白顆粒。
其生物學特性為:①宿主間可傳遞的傳染性,潛伏期可長達數年或數十年;②病理為變性改變,可見澱粉樣斑塊和膠質增生,但無炎性反應,亦無包涵體可見;③無復發緩解病程,持續進展直至死亡;④不產生干擾素不受干擾素影響孵育過程中不受任何免疫抑制或免疫增強的影響。
現在已知此慢病毒致病因子:①在高熱132℃ 60min可滅活;②亦可在10%次氯酸鈉溶液內浸泡1h以上或1N氫氧化鈉內浸泡30min,反覆3次即可滅活。
發病機制
 朊蛋白
朊蛋白PrP系一種單基因編碼的糖蛋白由253個胺基酸組成(鼠為254個胺基酸組成)位於人的第20號染色體短臂上,可譯框架由一個外顯子組成。在N的末端的附近由富有脯氨酸和甘氨酸的短肽5次反覆。正常中樞神經細胞表面也存在朊蛋白稱此為PrPc。分子量為30~33KD,其空間構象主要為α-螺鏇狀結構蛋白酶K可以溶解。而異常PrP被稱作為PrPsc,PrPES或PrPCJD。與PrPc截然不同。分子量為27~30KD其空間構象近40%為β層狀摺疊PrPSC數次的集結,則形成直徑為10~20nm,長度100~200nm的物質,這種物質可能就是早期發現的羊瘙癢病相關原纖維(scrapic-associated-fiber,SAF)和朊蛋白質粒(prion liposome)。它不能被蛋白酶K所消化。PrPSC大量沉積於腦內能摧毀自身的中樞神經系統,造成大腦廣泛的神經細胞凋亡、脫失,形成海綿狀腦病。
PrPSC是怎樣進入中樞神經系統,又是怎樣從正常的PrPc轉變為異常的PrPSC其詳細途徑和機制仍在研究中。不過,不同類型的CJD其發生機制也不盡相同;一般來說,醫源性CJD為傳遞感染,即將被PrPSC污染的組織或器械,通過腦深部電極檢查、顱腦手術、硬腦膜移植、以及反覆接受從垂體提取的生長激素或性激素肌注等,經過長達數年至數十年的複製而發病。家族性CJD則為PrP基因突變,即自體PrPc自發的發生結構改變,從而產生大量PrPSC導致中樞神經系統變性。而散發性CJD可能為體細胞突變的結果。
臨床表現
 腦幹
腦幹Gerstmann-Straussler-Scheinker(GSS)綜合徵是朊蛋白引起的家族性神經變性疾病,為常染色體顯性遺傳病變為小腦、大腦和基底核海綿狀變性顯著的澱粉樣斑塊沉積,合併脊髓小腦束和皮質脊髓束變性。
發病年齡19~66歲,平均40歲,發病及進展緩慢。病初主要表現小腦性共濟失調,最終合併痴呆、緩慢進展的痙攣性截癱,腦幹受累出現橄欖腦橋小腦變性症狀。病程持續2~10年EEG為彌散性慢波,無周期性改變。無特效治療。
2.致死性家族性失眠症(fatal familial insomnia,FFI) 的臨床特點
致死性家族性失眠症(fatal familial insomnia,FFI)是罕見的常染色體顯性遺傳病,至1998年為止僅發現9個家系23例FFI病人。朊毒體病由Lugaresi(1986)首先報告病變特點是丘腦變性
發病年齡18~61歲,病程7~36個月睡眠障礙是朊毒體病突出的早期症狀,病人總睡眠時間不斷地顯著減少,嚴重者一晝夜睡眠不超過1h,催眠藥無效。朊毒體病早期徵象也包括自主神經功能障礙可出現錐體束征小腦體徵痴呆和肌陣攣等。同一家族某些病人可出現CJD臨床表現,EEG可見彌散性慢波,周期性異常波罕見。FFI臨床表現多變,基因型檢查有助於診斷。朊毒體病無特效治療。
併發症
朊蛋白感染疾病的病變損害常累及皮質、底節、丘腦小腦、腦幹甚至脊髓前角等廣泛中樞神經系統導致精神、意識、智慧型障礙,故可以出現各系統功能失調及障礙,如延髓麻痹可致進食困難、嗆咳、肺部感染;腦幹病變也可影響心血管功能;長期癱瘓臥床引致褥瘡等。
診斷
 神經細胞
神經細胞1.多為中年以上發病。
2.既有神經症狀,如抽風共濟失調等又有精神症狀、記憶困難、智慧型低下、痴呆等
3.進展迅速 85% CJD在1年內發展為去皮質強直,GSS在2~3年內生活不能自理。
4.預後不良 CJD多於1年內死亡,GSS多於發病5年後死亡,FFI平均13.3個月死亡。
5.病理改變主要是神經細胞凋亡,星形膠質細胞增生和以灰質為主的海綿狀變性,嚴重者可累及白質但無任何炎症反應。
6.實驗動物可以傳遞。
鑑別診斷
朊毒體的鑑別診斷比較困難,腦活檢對臨床確診具有重要意義。
臨床診斷CJD時,應與Alzheimer病皮質下動脈硬化性白質腦病(Binswager病)、多梗死痴呆、多灶性白質腦病、進行性核上性麻痹、橄欖腦橋小腦萎縮、腦囊蟲、肌陣攣性癲癇等相鑑別。
檢查
1.血尿便常規、生化、肝腎功能常無異常所見。
2.腦脊液細胞和蛋白多數在正常範圍,少數病例蛋白輕度升高。雙向電泳可見異常蛋白利用免疫方法檢測腦脊液中14-3-3腦蛋白,對CJD具有極高的診斷價值。
3.血清S100蛋白的診斷價值 血清S100蛋白濃度測定,對CJD診斷特異性達到81.1%敏感性為77.8%。
其它輔助檢查:
1.腦電圖檢查 腦電圖改變被認為是臨床診斷CJD的重要根據,疾病的不同時期,腦電改變也不盡相同。
2.頭顱影像學檢查 通常在早期頭顱CT、MRI無異常所見。病情進展快至中晚期可見皮質萎縮,排除其他各種局灶性腦病,有助於臨床診斷。
3.正電子腦掃描(PET) 可測定大腦各葉代謝率變化。
4.腦活檢對臨床確診具有重要意義。
治療
朊蛋白感染疾病仍屬於無法治癒的致死性疾病,臨床只能對症處理併發症及給予支持治療。隨著人們對該病發病機制的逐漸闡明,不遠的將來人們有可能找到治癒這類疾病的原則與方法,能找到控制PrPC轉變為PrPSC或PrPCJD的途徑因為人們已經發現缺乏PrPC基因的鼠並不發生CJD,因此套用反義寡核甘酸或基因治療,可能達到預期目的。另一有利方面此組疾病的防治已引起WHO及其相關機構的高度重視。
預後:朊蛋白感染疾病潛伏期長、短病程,即發病後進展快,持續性進展,多在數月至1年內死亡,無特效療法。預後極差。
預防:預防重點應是嚴格處理朊蛋白感染疾病病人的腦組織血和腦脊髓以及與病人組織體液接觸或用過的手術器械、敷料及其廢棄物,要採取嚴格消毒措施。
手術器械可在高壓132℃ 60min或10%次氯酸鈉溶液浸泡60min,共3次。或1N氫氧化鈉溶液浸泡30min共3次。敷料和屍檢病理組織以焚燒處理為宜,取血注射器和針頭宜用一次性製品用後應作嚴格銷毀焚燒處理為妥善。醫護接觸病人尤。
