病因學
共濟失調微血管擴張症候群(Ataxiatelangiectasia;簡稱AT)是兒童時期最常發生的小腦運動失調(cerebellarataxia)的疾病,患者可能於3-6歲發病,並常合併免疫不全、微血管擴張,以及容易發生癌症,對離子化輻射導致細胞死亡的敏感度增加,並且對於輻射導致的DNA複製抑制現象,有不正常的阻抗現象等病徵。此症的發生是由於第11對染色體長臂23位置(11q23)上的ATM基因發生缺失所導致;ATM基因的蛋白質產物為一種proteinkinase,而這種kinase是細胞對DNA雙股螺鏇鏈斷裂時,產生反應的主要調節物質。此外也常可發現到患者發生染色體上的變異;約有5-15%的AT個案的lymphocyte在常規的染色體檢查中,發現在第7及第14對發生染色體的斷裂再重新接合的轉位情形。斷裂點的位置常發生在第14對染色體長臂11;該位置為T細胞α接受體,以及第14對染色體長臂32的位置上;該位置則與B細胞接受體的功能有關。此症為一體染色體隱性遺傳疾病,但與一般隱性遺傳疾病帶因者並無病徵不同,AT帶因者也常會發生輕微的臨床表征。
發生機率
此症為在多數國家中,最常在兒童時期造成進行性惡化的小腦運動失調的疾病,若合併動眼失用症的運動失調(ataxiawithoculomotorapraxia;AOA),可能於葡萄牙及日本人身上有較高的發生率。在美國的發生率約為1/40,000-100,000個活產新生兒。目前此症已列為政府公告之罕見疾病,據估中國發生率應為萬分之一以下。
遺傳模式
為體染色體隱性遺傳疾病,表示父母親為各帶一個突變的基因,下一代每一胎不分性別將有1/4的機率罹患此症。不同於一般隱性遺傳疾病帶因者的無病徵表現,帶有一個缺陷基因(heterozygote;異型結合子)的AT帶因者,常也有較高的罹癌機率,據研究統計約為一般人的4倍,其中以乳癌較為常見。
臨床表征
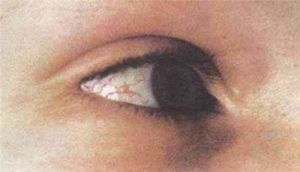
小腦運動失調為此症在症狀表現上的特徵及最主要的問題,症狀可能早在患者1-4歲就已發生,而出現步態及軀幹上的運動失調、舞蹈症(肢體紊亂的運動)、說話含糊不清,及動眼失用症的運動失調,而造成無法在視野中追蹤物體移動等病徵。動眼神經的微血管擴張(oculocutaneoustelangiectasia)則常於患者6歲時出現。患者常因免疫不全,而容易經常發生感染上的問題,此外患者易有發生癌症的傾向;最常發生為白血病(血癌)及淋巴癌。另一特徵為對離子化輻射(ionizingradiation)的有較高的敏感度。其它可能的病徵包括:提早老化(長出灰白髮)、內分泌異常等,偶有糖尿病的案例發生。常見的臨床表征詳述如下表:
| 小腦運動失調 | 若發生於學步後不久,患者開始會表現搖搖晃晃的步態。神經學上的症狀在2-4歲間可能稍有改善,短暫的改善可見於學習曲線的增長,但接下來將持續惡化。 運動失調的問題一開始發生於軀幹,但數年後也會影響到外圍肢體的運動協調,並開始出現口齒含糊不清,及視力上水平與垂直視野受限。 約25%的患者出現肌陣攣抽動及意向性顫抖;是指當患者意圖作某事時會抖得特別厲害,如:欲以手指觸某物時,在越接近目標物時抖得越厲害。 寫字的能力約7-8歲會受影響,多數患者10歲左右即可能需要以輪椅代步。 深腱反射減弱或消失,無法自抑的流口水。 肌肉的力量將逐漸衰弱至廢用,而發生遠程肢體的手腳指攣縮。 智力發展則正常不受影響。 |
| 神經病理學 | 小腦萎縮的情形,可能在患者7-8歲時,經腦部核磁共振檢查發現,並伴隨有小腦柏金氏細胞(Purkinjecells)及顆粒細胞(granulecells)衰亡的情形。 |
| 罹癌風險 | 發生癌症者約占AT患者的38%;其中85%為白血病(血癌)及淋巴癌;年幼患者較常發生急性淋巴球性白血病,較大的患者則可能為進行性T細胞白血病。淋巴癌則常為B細胞型。 隨患者的存活年齡愈長,也有發生其它如乳癌、卵巢癌、胃癌的案例。 |
| 免疫不全及感染 | 60-80%的AT患者會因免疫不全而發生易感染的問題,患者血中免疫球蛋白IgA、IgE及IgG偏低,約30%的患者T細胞不足。 多數研究發現,患者對使用肺炎鏈球菌疫苗的抗體反應不佳,死後解剖也常發現到患者與免疫相關的胸腺體積,都像胎兒時期一樣小。 不同於多數免疫不全的疾病,AT患者所感染的類型不包括伺機性感染。 有些個案可能出現慢性支氣管炎。 感染的嚴重度常與個案的營養及免疫狀況有關,免疫球蛋白的注射可能有助於改善經常感染的問題。 |
診斷
實驗室檢查
95%以上的患者血中α胎兒蛋白(alphafetoprotein;AFP)偏高,需注意的是少數一般正常小於24個月的嬰兒也可能會出現AFP偏高的情形。
免疫印跡法(Immunoblotting)
目前此技術為診斷此症最有力的工具,又稱為西方墨點轉漬法(Westernblotting),為檢測細胞或體液中特定的微量蛋白質。患者之中約90%檢測不到ATM蛋白,約10%的患者檢測得到極小量(traceamounts),1%的患者則有正常的ATM蛋白量,但無kinase活性(kinase-dead)。
輻射線敏感度試驗(Radiosensitivityassay)
此技術是在將實驗室中的轉殖細胞暴露於輻射線後,再進行轉殖細胞存活分析(colonysurvivalassay;CSA),多數患者(99%)的分析結果異常,但整個分析過程需3個月左右的時間。
染色體檢查
經過72小時PHA的刺激之後,部分AT患者的血液培養中,可能發現到第7對與第14對染色體轉位的變異。
基因檢查
以分子生物技術進行基因序列分析(Sequenceanalysis),約90%的患者可發現ATM基因的突變位置,不過仍有些在intro或heterozygous缺失無法偵測到。若未發現突變的ATM基因,對於有罹病風險患者家屬,可以基因連鎖分析(Linkageanalysis)比對是否與患者帶有相同的與疾病有關的基因片段。
治療
對於進行性惡化的運動失調的問題,目前尚沒有可以治癒或延緩的方法。可能可給予抗氧化劑的使用(如:維他命E、硫辛酸(alpha-lipoicacid)),但目前未有研究證實相關的治療成效。及早接受物理治療可使患者肢體攣縮的問題減到最小。免疫球蛋白可減少感染的次數,但通常較適用於經常發生嚴重感染的個案。由於患者對放射及輻射線的敏感度高於一般人;即使一般的劑量也有可能造成個案的死亡的風險,因此在進行相關檢查或治療時需格外小心。
預後
隨醫療技術的進步,在過去20年裡,AT患者的平均余命也較為提高,多數患者現今已能活過25歲以上,少數可至40-50歲。而可能合併有嚴重感染的肺臟衰竭則常為患者致命的原因。
