疾病概述
 埃萊爾-當洛綜合徵
埃萊爾-當洛綜合徵近年來,由於分子生物學及蛋白化學的發展,細胞外基質蛋白的主要成分及相應的多種基因突變已被確定。新技術的套用,如用轉基因動物模型觀察基質基因產物的基本功能,包括轉錄因子、生長因子、分化因子和細胞因子等,使發病機制的研究更加深人。目前的難點是認識特殊基因突變與臨床表現型的關係,突變的病理機制等以便制定合理的臨床治療策略
1.膠原生物合成的分子調控與臨床疾病
(1)膠原的結構與功能:膠原保持某些器官正常的結構及功能,如眼、心肌、心瓣膜、骨骼肌、韌帶、肌腱、腎、關節、軟骨等。原膠原由3條多肽鏈(alpha鏈)組成,其胺基酸順序為GLY-X-Y,GLY為甘氨酸,占1/3,X多為脯氨酸,Y常為羥脯氨酸占1/4,3條鏈之間由氫鍵相連成3螺鏇,在兩端有N及C末端前肽。目前已確認的膠原分19型,編碼30個基因,分布於12條染色體上。由於轉錄不同的基因剪下片段或用不同的引物轉錄的不同RNA,使蛋白的排序多樣化。膠原蛋白被小膠原(minor collagen)或非膠原蛋白調控原始蛋白被特殊的或小膠原修飾,組裝成適應一些特殊需要的結締組織如抗牽拉、抗壓力及屏障作用細胞外基質蛋白(ECP)合成缺陷導致相關疾病
(2)結締組織的功能:
①Ⅰ型膠原家族的作用:Ⅰ型膠原家族的作用是保持皮膚,肌腱,韌帶的張力在Ⅰ型膠原纖維內有小量Ⅴ型膠原;Ⅶ型膠原分布在纖維束的表面,Ⅵ型分布在基質內,有助於間質膠原的固定。這些組織與非膠原蛋白之間的相互作用形成膠原家族結構的多樣性。基質的蛋白多糖中的核心蛋白聚糖(decorin)附著於膠原,功能是固定變形的生長因子-β分子到纖維表面。非膠原基質蛋白如磷酸蛋白及骨鈣素(osteocalcin)使骨堅固。Ⅲ型膠原蛋白是內臟平滑肌細胞的結締組織。Ⅰ型膠原與彈力組織的相互作用可約束血管壁。
②抵抗壓力作用:Ⅱ型膠原是軟骨的主要組成部分。其纖維由Ⅺ膠原及Ⅸ型膠原調控特殊的軟骨組織如肥大軟骨細胞由Ⅹ型膠原產生;Ⅵ型膠原分布在軟骨,使之與周圍結構固定,Ⅱ型膠原分布於關節面鼻、耳、眼玻璃體。
③各型細胞間的屏障及相互溝通作用:結締組織的另一功能是維持各型細胞間的屏障機制及相互溝通。此功能主要靠基底膜的濾過作用,基底膜主要由Ⅳ型膠原組成。Ⅶ型膠原的功能是將基底膜固定在臨近組織。Ⅷ型膠原主要出現在血管,神經組織。這3種膠原保證多種組織如角膜、血管內皮腎小球的基底膜等的功能正常
新分類的膠原分子如ⅩⅤ及ⅩⅦ型膠原可能有細胞與細胞外環境之間通道的作用。細胞外基質可通過細胞膜受體接受信號,合成所需要的成分,以適應組織生長及修復。
(3)膠原生物合成與臨床疾病:由Ⅰ型膠原合成途徑缺陷導致的疾病的研究成為所有原纖維膠原突變的例證,有助於對更複雜的突變進行更深入的研究。由於膠原基因突變,或由於介導翻譯後的膠原蛋白及細胞外基質代謝的酶缺陷所致的疾病有多種,如成骨不全(osteogenesis imperfacta)軟骨發育不全(achondroplasia)、埃萊爾-當洛綜合徵(Ehlers-Danlos syndrome)、X-連鎖.Alport綜合徵(Alport's syndrome)大皰性表皮裂解(epidermolysis bullosa)等。
(4)膠原合成(圖1):
①膠原基因在不同細胞中的多種表達:Ⅰ型膠原基因大而複雜,分布於50或51個內含子範圍膠原基因的表達水平取決於其所含的對轉錄因子有不同反應的DNA原件(element)上的啟動子(promotor)這些原件定位於基因編碼區的遠端(5上游)及內含子序列內主要在骨組織表達的DNA原件與在肌腱,血管平滑肌,皮膚的不同,說明一個單個基因在不同細胞中的多種表達。
②轉錄:原始膠原信使RNA(mRNA)的轉錄是含內含子,外顯子基因的完全複製。Ⅰ型膠原的雜合二聚體(heterodimer)[α1(Ⅰ)]2α2(Ⅰ)從每個基因成倍轉錄。轉錄後的核mRNA進入加工程式,移出內含子因為內含子序列會改變RNA閱讀框架或使不適合的胺基酸進入編碼蛋白以及使一些異常產物保留在細胞核內並被降解,使正常的mRNA產物減少故內含子必須移出。此過程是在內含子外顯子交接點出現識別序列(為一組小的核RNA(nuclear RNAS)),經剪下,將全部內含子序列移出,使相鄰外顯子連線。mRNA到達粗內質網後膠原被翻譯成多肽α(alpha)鏈。mRNA原始序列的異常如終止密碼的1個鹼基改變,或閱讀框架的移位都會使蛋白產物減少。
③加工及鏈裝配:膠原mRNA被加工某些脯氨酸殘基羥化賴氨酸羥化、糖基化形成胞質mRNA。羥化的脯氨酸殘基使膠原三螺鏇在生理溫度條件下更穩定。脯氨酸羥化酶已被克隆其活性與膠原合成的速度平行,賴氨酸的羥化使骨組織形成穩定的中間鏈及交叉連鎖(cross-links)基因突變可導致的過量賴氨酸羥化會影響三螺鏇形成。
④胞質mRNA從多肽C端向N端自我裝配成三螺鏇,形成細胞內前膠原。並分泌到細胞外,此過程在高爾基體進行。
在細胞外,C及N端的多肽從裝配好的細胞內前膠原(intracellular procollagen)移出,形成細胞外膠原(extracellular collagen)。所有膠原α鏈C端都有一高度保守區,對鏈裝配非常重要。此區分子突變會使異常鏈進入三螺鏇,導致膠原形成減少。GLY-X-Y三體的第1位的甘氨酸殘基其功能是使多肽鏈保持緊的結構,如發生點突變可導致的甘氨酸替換影響三螺鏇形成,裝配慢,分泌差,對組織蛋白酶敏感,影響正常功能。
⑤微纖維(microfibril)形成及交叉連鎖,形成成熟的膠原:形成成熟的膠原纖維的最後步驟是個體分子進入膠原多聚體,隨後,分子間交叉連鎖使分子內穩定,此過程由賴氨酸氧化酶啟動,由三螺鏇外露區的信息指導,最後形成不溶性的膠原(insoluble collagen)。微纖維的正常排列對賴氨酸氧化酶啟動交叉連鎖至關重要。突變導致的微纖維排列紊亂,使膠原交叉連鎖發生缺陷會弱化結締組織阻斷交叉連鎖的形成的物質如青黴胺會增加組織脆性致骨彎曲,動脈瘤等。賴氨酸氧化酶基因已克隆,定位在5號染色體。交叉連鎖的形成的遺傳缺陷尚無報導。放射免疫法測定這些前肽,對估價一些疾病的膠原合成率對激素治療的反應有臨床價值。
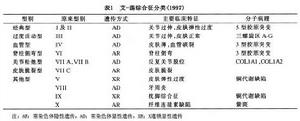
2.現將埃萊爾-當洛綜合徵分類歸納於表1。
埃萊爾-當洛綜合徵(Ehlers-Danlos syndrome,簡稱EDS)又稱全身彈力纖維發育異常症。臨床以皮膚及關節過度伸展,組織易於損傷,脆性增加及創傷不易癒合,血管脆性增加,眼部異常及內臟器官異常為特徵,是結締組織主要蛋白質之一的膠原先天性代謝異常症根據臨床特點與遺傳學特徵,可以將本病分為11個亞型。本綜合徵最早由:Ehlers(1901)和Danlos(1908)提出。
病因
本病病因目前尚不十分清楚,一般認為是中胚層細胞發育不全致膠原蛋白轉錄和翻譯過程缺陷或翻譯後各種酶缺陷使其合成障礙而引起。
發病機制
 埃萊爾-當洛綜合徵
埃萊爾-當洛綜合徵本病發病機制目前還不十分清楚。多有家族史,發病多符合常染色體顯性遺傳或常染色體隱性遺傳,部分符合X連鎖隱性遺傳。
Ⅰ、Ⅱ、ⅢⅧ、Ⅺ型為常見染色體顯性遺傳臨床研究證明,膠原蛋白Ⅰ的α-1和α-2基因定位於7號染色體。Ⅰ、Ⅱ、Ⅲ型膠原蛋白即問質膠原蛋白,由基因組合顯示,沿著三螺鏇區域的進化保守位置有大量相對小的外顯子,使其Ⅰ型膠原的可溶性增加,超微結構示膠原纖維直徑增加,交聯異常。前膠原肽酶缺損,可能是因膠原的一級結構異常或代謝異常所引起。常染色體隱性遺傳者,其皮膚或大動脈等組織中無Ⅲ型膠原,即使在成纖維細胞培養時也無Ⅲ型膠原合成因此認為可能是由於與Ⅲ型膠原相關基因異常所引起1990年Zafarullah等證實了在三螺鏇531位胺基酸密碼子GCT(丙)→ACT(蘇)的改變其丙氨酸等位基因的頻率為0.68Ⅲ型膠原同Ⅰ型膠原一樣幾乎遍布全身,尤其是動脈中層、大動脈內膜和肺泡隔等間質處,主要由Ⅲ型膠原構成。根據其分布部位分析,Ⅲ型膠原可能與某些組織彈性的穩定性有關另外對纖維形成起作用的Ⅰ型膠原也有很大影響。因此,Ⅲ型膠原缺損可出現因血管和各種臟器強度減弱而引起的各種臨床症第Ⅳ型通常為常染色體顯性遺傳,但也有常染色體隱性遺傳或性連鎖隱性遺傳。本型膠原蛋白基因定位於164q21~q31。1988年Superti-Furga等證明成纖維合成了正常大小的和縮短的Ⅲ型膠原鏈,在三螺鏇區域內其基因和中部有一大的缺失即編碼區外顯子16缺失。1991年Richards等發現纈氨酸替換甘氨酸910的G→T突變。1992年Kontusaari等發現COL3-AⅠ基因的單個鹼基替換,將1018位的甘氨酸密碼子轉變成天冬氨酸密碼子,由於甘氨酸的突變,使其皮膚成纖維細胞分泌到介質中的Ⅲ型膠原蛋白量顯著降低。在有些病例其成纖維細胞可以合成Ⅲ型膠原前質,但其全部膠原前質向細胞外的分泌障礙。
第Ⅴ型為性連鎖隱性遺傳Ⅴ型膠原蛋白基因定位於2q24.3~q31。本型膠原蛋白有3個鏈的變異體它有特異性細胞周圍的分布,通常位於基底膜和間質之間,可能有助於大直徑纖維的定向。在真皮成纖維細胞培養合成的膠原易溶同時在細胞與培養液中的賴氨醯氧化酶活性降低。此種酶與膠原及彈性蛋白的交聯形成有關,故此酶缺乏可使正常膠原纖維交聯形成受抑制,導致膠原纖維形成障礙。
第Ⅵ型為常染色體隱性遺傳,本型膠原蛋白基因定位於2q27.3。病人皮中羥賴氨酸殘基減少,尿中羥賴氨酸排泄亦減少。另外成纖維細胞培養時,賴氨酸羥化酶活性降低。羥賴氨酸對Ⅰ型膠原的交聯形成有特別重要的作用,它的缺損可致富有Ⅰ型膠原的皮膚膠原缺乏交聯,從而減弱皮膚彈性的穩定性,引起各種臨床症狀。該亞型主要因Ⅰ型膠原酶缺損所引起。
第Ⅶ型多為常染色體隱性遺傳,本型膠原蛋白基因定位於3p21.3Ⅶ型膠原蛋白有1個三螺鏇區域,它較Ⅰ型蛋白的三螺鏇區長一半,並以二硫鍵穩定的二聚體分布於基底層之下的真皮-表皮基底膜區,造成膠原前質異常堆積於皮膚等結締組織中,使正常膠原成熟發生障礙。Ryynanen等1992年證實Ⅶ型膠原蛋白在真皮-上皮基底膜區表達,在人類皮膚發育過程中可能是該型膠原蛋白的主要細胞來源。Ⅶ型膠原蛋白僅局限於分層的鱗狀上皮之下的基底膜區在該皮膚基底膜區內,本型膠原蛋白位於上部乳頭真皮的緻密層和亞緻密層區域內。免疫定位證實本型膠原蛋白是錨定纖維的主要膠原組成成分在對真皮胺基酸進行分析證明,其胱氨酸顯著增加,甘氨酸、羥脯氨酸減少,非膠原成分增加,患者成纖維細胞的氨基端膠原前質肽酶活性明顯下降,表明該酶缺損可使膠原前質合成過多而引起相應的臨床症狀
第Ⅷ型為常染色體顯性遺傳,本型膠原蛋白基因定位於在3q12~q13.1。在膠原蛋白中該型膠原蛋白因其組織分布和生物合成性質而可能是獨特的Ⅷ型膠原蛋白-N-原肽酶裂解位點和正常時參與膠原纖維內共價分子間交聯的一個賴氨酸殘基的丟失,致使在內皮細胞基底膜的主要成分減少。
第Ⅸ型Ⅹ型為常染色體隱性遺傳,前者膠原蛋白基因定位於6q12~q14後者則定位於6q21~q22.3Ⅸ型膠原蛋白含有半胱氨酸殘基的短非膠原肽,為軟骨特異性膠原蛋白。Ⅹ型是軟骨的一短鏈次要膠原蛋白,在長骨的生長和發育期間,軟骨細胞依次經過增殖期、肥大期和變性期,形成了軟骨發育不良和軟骨性的其他疾病,大部分可有血小板功能的改變
此外,由於膠原蛋白亞型的不斷出現,其新的分型和基因定位也相繼被標明,如Ⅻ型膠原蛋白基因定位於6q12~q24;ⅩⅤ型膠原蛋白基因定位於9q21~q22;ⅩⅥ型膠原蛋白基因定位於1q34~q13;ⅩⅦ型膠原蛋白基因定位於6號染色體Ⅷ型膠原蛋白基因定位於21a22.3。通過連鎖分析膠原蛋白的三核苷酸結構重複序列不穩定,有可能導致諸如無法解釋的膠原性疾病或可疑性膠原病。
病理:利用組織學、組織化學和電鏡檢查患者皮膚及其他器官的彈力纖維,We-schler發現膠原纖維量減少,彈力纖維減少。Gulkumen觀察病人的皮膚膠原束排列紊亂,其大小也有改變,而且彈力纖維交織網增多。
血管病變嚴重的病例,可見有動脈彈力纖維破裂成碎片及黏液性水腫性退行性變總之,不同報導者報導的病理改變亦不相同。
臨床表現
 埃萊爾-當洛綜合徵
埃萊爾-當洛綜合徵常為早產兒,多伴有胎膜早期破裂嬰兒則表現為肌張力低下
1.本綜合徵的共同特徵有
(1)皮膚和血管脆弱,皮膚輕度損傷即易撕裂傷口癒合慢,皮下血管脆性增加,輕傷也易形成瘀斑。
(2)皮膚過度伸展可牽引出很長的皮襞,老年時皮膚松垂,尤以肘部為著全身皮膚變薄。
(3)關節活動範圍過大,髕肩、髖、鎖骨及顳頜關節易脫位幼兒關節活動過度則易摔跤,患者可以自動或被動伸展。
(4)束臂試驗陽性。
(5)常伴有繼發感染
(6)有時合併心臟畸形如二尖瓣脫垂,主動脈弓異常,雙瓣型主動脈瓣,肺動脈狹窄,房室間隔缺損,法洛四聯症等
(7)其他:可發生各種疝如臍疝、腹股溝斜疝、裂孔疝等,肺部病變如肺破裂、氣胸、肺氣腫等齲齒或牙周炎也可發生。
2.本綜合徵及其11個亞型分別介紹如下
(1)第Ⅰ型:又稱Gravis型最多見也稱重症型早產兒多見,由於胎膜主要來自胎兒,其結締組織的脆性增加故胎膜早期破裂。新生兒可有先天性髖關節脫位,幼兒時由於關節活動過度難以控制經常摔跤隨著小兒的生長發育,可發生選擇性關節習慣性脫位、關節腔積液、足畸形、腰椎畸形。常發生靜脈瘤。
(2)第Ⅱ型:又稱Mitis型本型為第Ⅰ型的輕型,關節活動過度多見,還可有手掌的皮膚鬆弛足底常有皺褶皮膚脆性增加,創傷後傷口癒合慢。本型較Ⅰ型症狀輕。
(3)第Ⅲ型:本型又稱良性過度活動型。該型以關節活動過度為其特徵,主要表現於髕肩、髖、鎖骨等關節異常,合併慢性脫位。皮膚骨骼畸形較輕。
(4)第Ⅳ型:為皮下出血型動脈型或稱Sack型,無皮膚過度伸展,但皮膚菲薄,可以透見皮下的網狀靜脈,易於出血。常因動脈破裂或消化道穿孔而死亡,很少活到20歲。有時可合併多種先天性心血管系統異常包括法洛四聯症、房間隔缺損肺動脈和主動脈異常等。此型病人可形成主動脈夾層動脈瘤而且血管瘤易於自發性破裂也可出現主動脈根部擴張而造成主動脈瓣反流。
(5)第Ⅴ型:皮膚過度伸展症狀同第Ⅰ型,但其關節過度活動性較輕且局限常有骨關節畸形及關節血腫身材多矮小易合併先天性心臟病,尤以二尖瓣脫垂為多見。
(6)第Ⅵ型:除有本綜合徵上述的共同特徵外,尚有圓錐角膜、晶體脫位、視網膜剝離、蜘蛛狀指、脊柱側彎等馬方綜合徵樣症狀。因此,本型也稱馬方樣過度活動綜合徵
(7)第Ⅶ型:亦稱多關節鬆弛型,以多關節鬆弛為其主要臨床表現可出現反應遲鈍
(8)第Ⅷ型:為牙周炎型,本型患者皮膚脆性增加,易創傷後出血,關節活動過度僅限於手指臨床表現以牙周炎為其特徵。
(9)第Ⅸ型:本型多表現為智力低下除皮膚過度伸展和皮膚脆性增加外,多有嚴重的智力低下並有疝的形成,如臍疝、腹股溝斜疝等。
(10)第Ⅹ型:為血小板功能障礙型本型除埃萊爾-當洛綜合徵共有的特徵外,其特異性表現為血小板聚集功能障礙。
(11)第Ⅺ型:為關節鬆弛型,本型以關節活動過度為其特徵,尤以肩關節脫臼為常見。
現將各型的臨床特徵歸納於表2
除上述症狀外,埃萊爾-當洛綜合徵還可出現神經肌肉症狀中樞神經系統症狀主要由腦內動脈瘤引起,如腦動脈瘤破裂引起嚴重的蛛網膜下腔出血,頸內動脈海綿竇瘺造成的壓迫症狀,視網膜血管迂曲、擴張引起的增生性視網膜炎或視網膜剝離及癲癇大發作等。肌肉症狀可見有肌肉發育不良伴肌無力,也可見有肌萎縮。
併發症:
1.第Ⅰ型 可並發選擇性關節習慣性脫位、關節腔積液、足畸形、腰椎畸形常發生靜脈瘤
2.第Ⅲ型 可並發,髕、肩、髖鎖骨等關節異常,合併慢性脫位。
3.第Ⅳ型 可並發出血。常因動脈破裂或消化道穿孔而死亡。
4.第Ⅴ型 可並發骨關節畸形及關節血腫,易合併先天性心臟病尤以二尖瓣脫垂為多見。
5.第Ⅸ型 多並發嚴重的智力低下,並有疝的形成,如臍疝、腹股溝斜疝等。
診斷
根據皮膚和血管脆弱,皮膚過度伸展,關節活動範圍過大3大主症,即可診斷為本綜合徵,再結合其他器官或系統的異常發現,可具體確定其亞型。
鑑別診斷:
必須與下列疾病加以區分:
1.馬方(Marfan)綜合徵 大多同時出現骨骼異常如肢體細長、蜘蛛指等,眼部異常可出現晶體脫位、青光眼及高度近視等,心血管系統異常可出現升主動脈進行性擴張,伴主動脈瓣關閉不全肺動脈突出等。另外尚可有特殊面容,即長方頭、狹長臉、上齶弓形高聳等。雖有關節鬆弛過伸,但無皮膚脆弱過伸症狀。
2.彈性假黃瘤病 對稱性分布於皮膚皺褶部位,呈簇狀或網狀小的淡黃色斑塊或小結節皮損,以及皮膚鬆弛、四肢血管供血不全、脈搏異常心肌炎與主動脈炎等心血管異常,眼底有血管樣色素紋特徵性改變,但無關節鬆弛表現。
3.皮膚鬆弛症 本病主要表現為皮膚鬆弛尤其在大皺襞處可見鬆弛的皮膚明顯懸垂或早老現象,一般無關節活動過大。此症皮膚用手捏起再放鬆時,其回縮力很差。本病與埃萊爾-當洛綜合徵的Ⅸ型較難區別,均有賴氨酸氧化酶缺乏但二者遺傳方式不同,本病多見於常染色體顯性遺傳。
檢查
 埃萊爾-當洛綜合徵
埃萊爾-當洛綜合徵實驗室檢查:
1.血液學檢查 多次消化道出血者,可有不同程度的貧血、血小板減少、某些凝血因子異常及束臂試驗陽性。
2.免疫學檢查 常見有IgA、IgG或IgM降低及E-玫瑰花結形成數降低。
其它輔助檢查:
X線檢查。
1.顯示 皮下有散在的小圓形鈣化結節,且兩側常呈對稱性排列,最常見於肢體伸側。下肢的結節可見於內、外側,結節中心為透光區,四周環繞濃密陰影也可為瀰漫性或斑點狀鈣化。
2.肘、膝關節間隙增寬 半脫位或脫位骨質疏鬆甚至可有肢端骨質溶解骨骼發育和顱骨骨化延遲
3.骨骼其他異常 有尺骨莖突長,尺橈骨骨性聯合第5指近節指骨短,畸形足多余齒,脊椎後凸以及各種胸部異常等。
4.心血管造影 可顯示主動脈狹窄、主動脈瓣閉鎖不全、二尖瓣關閉不全、動脈主幹自發性破裂夾層主動脈瘤、動靜脈瘺及其他先天性心臟畸形。
治療
本綜合徵無特效療法,輕症者不需治療,重症者可予對症處理。如有動脈瘤可進行手術治療,有多動症狀者可用中樞興奮藥呱酯甲酯。
1.內科對症治療 消化道出血時應給予輸血和止血藥物;心力衰竭者可給予強心藥物及利尿藥物;合併感染時,可用抗生素。
2.手術治療 一旦發生關節脫位,則需予以及時復位,並用繃帶固定數周。對於脊柱畸形,可行矯形手術,但需注意預防手術併發症如出血過多、縫合及癒合不良等。有心臟瓣膜畸形者,可行瓣膜置換手術。
預後預防
 埃萊爾-當洛綜合徵
埃萊爾-當洛綜合徵預後:
無心臟病變者,預後良好。有心臟病變者多死於心力衰竭。第Ⅳ型 為皮下出血型、動脈型或稱Sack型無皮膚過度伸展,但皮膚菲薄可以透見皮下的網狀靜脈,易於出血。常因動脈破裂或消化道穿孔而死亡,很少活到20歲。
預防:
1.一級預防 遺傳病的預防,除了從整個人群的角度做好流行病學調查、攜帶者檢出、進行人群遺傳監護和環境監護,開展婚姻和生育指導,努力降低人群中遺傳病發生率,提高人口素質之外,針對個體,必須採取有效的預防措施,避免遺傳病後代的出生(即實行優生)和遺傳變異的發生,採取通常的措施包括:婚前檢查、遺傳諮詢、產前檢查和遺傳病的早期治療。
(1)婚前檢查:婚前檢查(即婚姻保健),它是保證男女雙方婚後生活幸福、後代健康的重要環節。婚前檢查的重點是:①遺傳病方面的調查,包括詳細詢問男女雙方及其家庭成員的健康狀況既往病史及醫治情況,尤其是有無先天畸形,遺傳病史和近親婚配史。必要時應進行家系調查、血型檢查、染色體檢查或基因診斷,以檢出攜帶者;②全面的體格檢查,主要是對急性傳染病結核病,或嚴重的心、肝、腎疾病泌尿道慢性炎症等可嚴重威脅個人或配偶健康的疾病,以及女方的嚴重貧血、糖尿病等可對胎兒造成影響的疾病的檢出,並動員經治癒後才可結婚;③對男女生殖器官的檢查,檢出性器官畸形兩性畸形等疾患,以便極早採取措施。
(2)遺傳諮詢:遺傳諮詢(genetic counselling)是由臨床醫生和遺傳學上,作肯解答遺傳病患者及其親屬提出的,有關遺傳性疾病的病因、遺傳方式、診斷、治療及預後等問題,估計患者的子女再患某病的機率,並提出建議及指導,以供患者及其親屬參考。遺傳諮詢的意義在於:①減輕患者身體和精神上的痛苦減輕患者及其親屬的心理壓力,幫助他們正確對待遺傳病了解發病機率,採取正確的預防、治療措施;②降低人群遺傳病的發生率,降低有害基因的頻率,及減少傳遞機會。
①遺傳諮詢的分類和內容:
A.婚前諮詢:婚前男女雙方在得知一方或其親屬中有某種遺傳病時,詢問能否結婚?後代中該病的發病情況怎樣?
B.產前諮詢:夫妻中的一方或其親屬中有某種遺傳病或先天性畸形詢問後代類似疾病的發生情況;若已作過某種遺傳病或先天性畸形,詢問再生育時後代情況及如何預防患兒的出生。妊娠期曾患某病、服用某藥或接觸有毒物質或放射線詢問胎兒的可能情況。
C.一般遺傳諮詢:除了上述的情況外,還詢問可否近親婚配?已經發病個體的防治辦法出現某些症狀或體徵疑為遺傳病者想得到解釋等
儘管諮詢者的年齡、職業知識基礎和文化水平各不相同,來意和要求也不一樣,但遺傳諮詢的基本內容可以歸納為以下4方面:a.明確診斷是否屬於遺傳性疾病;b.解答各種問題,包括防治和預後;c.推算復發風險率;d.商討對策。為了完成這些內容,遺傳諮詢通常採用的程式包括:a.通過病史和家系調查,體格檢查和必要的輔助檢查和特殊的遺傳學檢查分析,確定是否為遺傳病,以何種方式遺傳;b.按照遺傳方式和特點估計復發風險率;c.通過商談討論、提出預防、治療對策和婚姻、生育指導。
②復發風險率的估算:
A.人類遺傳病的復發風險,按其危險程度可分為3類:
a.一般風險:發病幾率為1∶20以上。常指由於環境因素等(如孕婦妊娠初期感染風疹等)而引起的疾病,它對以後的同代個體的發病一般無影響,預期風險近似整個群體的風險。
b.輕度風險:發病機率1∶10~1∶20。常指多基因遺傳病的復發風險,要根據該病的遺傳度、閾值等進行綜合分析計算
c.高度風險:發病幾率為1∶1~1∶10。所有單基因遺傳病(常染色體顯性遺傳病,隱性遺傳病,X伴性遺傳病)以及雙親之一具有平衡易位染色體的情況,均屬此類。
B.遺傳病的復發風險率估算:隨不同遺傳方式的遺傳病和已知信息的不同而定。在單基因的復發風險估算中分基因型已推定者和基因型未能推定者兩種情況進行估算。
a.基因型已推定者:常染色體顯性遺傳病因患者多為雜合體,若外顯率為100%雙親一方為患者,則子女得病機率為50%若已生育1個或幾個患者,復發風險仍為50%,雙親均為患者時子女的復發風險為75%,雙親皆非患者的子女,一般不發病。突變個體的子女復發危險率為50%,其同胞發病率等於群體自然突變率若外顯不全,則子女得病機率為K/2(K為外顯率,即實際觀到的患病數與預期值的百分比)。
常染色體隱性遺傳病:子代發病風險與雙親的情況關係密切(表3)。若是近親婚配子女患病風險提高。
X伴性顯性遺傳病:男性患者與正常女性婚配其子女中男性正常女性發病。男性正常與女性患者婚配的子女各有50%發病。
X伴性隱性遺傳病:男性患者的兄弟有50%可能發病,其姐妹不發病,但有50%是攜帶者,同胞中總發病率為25%;其子女一般都不發病,但女兒為攜帶者,女性患者的子代中男孩100%發病,女兒都是攜帶者。男性患者與女性攜帶者婚配,其子女中各有50%發病女性攜帶者與正常男性婚配,則子女中男孩有一半發病,女孩有一半為攜帶者。
Y伴性遺傳病:一般表現為父傳子,子傳孫僅限男性發病。
b.基因型未能推定者:若雙親之一或雙方的基因型未知,則估計發病子女或以後出生子女復發風險複雜得多。因為遲發外顯性遺傳病雜合體要到一定年齡才發病一個健康子女可以是完全正常的也可能是一個尚未發病的雜合體要估計復發風險,就必須推定其為雜合體的機率。在隱性遺傳病家族中,表型正常的雙親,生下正常的小孩,也不能斷定他們不是遺傳攜帶者,因為,即使雙親都是雜合子,生育正常小孩的機率也有3/4。當然,他們生下正常小孩越多,是雜合子的機會越小在這種情況下,估計復發風險,可根據上代和下代的表現型實驗事檢查的結果用逆機率定律(Bayes定律)來推算。
多基因遺傳病的致病基因有多對,呈共顯性;每個基因作用微小,但具有累加效應,而且,除了遺傳基礎外,環境因素在多基因遺傳病的發病中起較大的作用。不同的多基因病的遺傳度不同發病閾值也有差異推算復發風險率時,比較複雜。一般來說,遺傳度比較高(70%~80%)的多基因病,若群體發病率為0.1%~1%,則患者一級親屬的發病率近似於群體發病率的平方根除此以外。還應考慮幾個問題現舉例加以說明。
 埃萊爾-當洛綜合徵
埃萊爾-當洛綜合徵唇裂在我國的群體發病率為0.17%,遺傳度為76%,患者一級親屬發病率為4%,接近於0.17%的平方根;當1對夫妻生了2個唇裂患兒後,復發風險率就相應增加,發病率由4%增至10%;如果患者病情嚴重,其發病風險率比病情輕的將增高一側唇裂患者的一級親屬發病率為2.6%,而兩側唇裂並齶裂者,復發風險率可高達5.6%當發病率有性別差異時發病率低的某性別患者的一級親屬復發風險率比發病率高的性別患者的一級親屬的復發風險率要高;先天畸形在新生兒中占1%~2%,當已出生1個這類畸形兒時,再次妊娠發生這類畸形的風險率即隨已有患者數增多而增高
多數染色體病患兒的親代核型都正常,由於生殖細胞發生過程出現染色體畸形而導致後代發病這類患者的同胞再患該病的風險率與一般人群相同;高齡產婦或有明顯誘變因素接觸史的雙親,復發風險率可顯著增高;染色體數目異常(如13、18、21等染色體的三體性綜合徵),如果雙親之一的核型為嵌合型則再生患兒的風險率可用下式估計P=[X/(2-X)]÷K(P為風險率,X為三體細胞的百分數,K為係數,通常為2),染色體結構畸變所致疾病的復發風險率的計算,需根據不同的畸變類型,分析可能出現的分離、交換形式最後根據分離定律和交換定律分析配子的情況,才能得到估算。
③提出對策和解答問題:遺傳諮詢工作在詳實地了解病史及家系情況,分析了遺傳方式和估計了復發風險率後,要對患者及其家屬提出的問題加以解答對遺傳病的治療、預防等問題提出對策,並給予婚姻、生育等方面的指導。以切實起到預防遺傳病發生,造福人類的作用。
(3)產前診斷:產前診斷(prenatal diagnosis)又稱為宮內診斷(intrauterine diagnosis)或出生前診斷(antenatal diagnosis)是通過對孕期胎兒性別及健康狀況的檢測,以便及時採取必要的措施防止遺傳病或先天畸形患兒的出生產前診斷是生化遺傳學、細胞遺傳學、分子遺傳學和臨床實踐相結合的產物,當今高解析度顯帶技術基因工程技術以及絨毛吸取和培養技術的發展使產前診斷套用面更廣,檢查結果更準確。
①產前診斷的對象:
產前診斷常遇到的遺傳病有下列3類:
第1類:染色體異常約占出生總數的0.5%,由於容易明確診斷。故可占產前診斷病例的1/4~1/2。
第2類:單基因病,一般占出生總數的3.5%,占產前診斷病人的10%左右。
第3類:多基因病,包括無腦兒、脊柱裂、腦積水某些唇裂和齶裂及某些先天性心臟病等。主要是神經管畸形,占產前診斷病例的40%~50%。
產前診斷適應證的掌握不同國家、不同醫院有些區別,一般衛生保健條件好的地區和醫院掌握較寬。通常普遍接受的適應證包括以下幾條:
A.35歲以上的高齡孕婦。
B.夫婦一方有染色體數目或結構異常的孕婦。
C.已生過21三體綜合徵或其他染色體異常患兒及有相應家族史的孕婦。
D.具有脆性X染色體家系的孕婦。
E.夫婦一方是染色體平衡易位或其他染色體畸變的攜帶者或嵌合體的孕婦。
F.夫婦一方是某種遺傳病患者,或曾生育過某種遺傳病患兒的孕婦。
G.夫婦一方有神經管畸形,或生育過開放性神經管畸形兒(無腦兒、脊柱裂)的孕婦。
H.有原因不明的自然流產,死產、新生兒死亡等病史的孕婦。
I.在妊娠早期曾受較大劑量的輻射或受病毒感染、長期服藥的孕婦。
J.羊水過多的孕婦。
K.夫婦一方有明顯的環境致癌致畸、致突變因素接觸史的孕婦。
②產前診斷的方法:產前診斷按檢查對象不同,可以分為母體篩檢和胎兒檢查。其中母體篩檢可以有母體血血清甲胎蛋白篩檢、母體循環血中胎兒細胞檢查等。而通常所指的產前診斷,主要是對胎兒的檢查診斷。它可以在不同水平上進行,選用不同的方法
A.形態學水平(表型水平):主要檢查胎兒有無先天畸形,常用的手段有:
a.X線檢查:妊娠16周后,胎兒四肢長骨、短骨、肋骨等都已經骨化可以通過X診斷畸形。在必要時,可用水溶性或油溶性造影劑注入宮腔,進行羊膜腔造影。
b.超聲診斷:超聲診斷是一種簡便而且損傷極小的產前診斷方法。常用的超聲診斷儀有A型超聲診斷儀B型超聲診斷儀,超聲都卜勒診斷儀和M型超聲診斷儀B型超聲診斷儀(B超)具有光點反差大,圖像清晰,分辨力高等優點多探頭電子自動快速掃描,提高了掃描速度可以直接觀察胎心。胎動等動態,並可攝像記錄分析。B超常用於檢出:多胎妊娠;胎盤定位;性別鑑定;神經管畸形;內臟畸形;胎兒有核紅細胞增多征;胚胎髮育異常;宮內生長遲緩。
c.胎鏡:胎鏡(fetoscope)是一種帶有羊膜穿刺的雙套管的光導纖維內鏡。插入羊膜腔後,可直接觀察胎兒畸形,也可採集胎兒活體組織和胎血絨毛等材料還能開展某些宮內治療為遺傳病的預治,提供了新途徑但此操作可引起流產、羊膜炎、母體免疫反應等併發症,故其套用受到一定的限制。
B.染色體水平:對宮內胎兒作染色體檢查,能早期診斷和預防常見的染色體病、脆性X染色體綜合徵、染色體斷裂綜合徵以及與染色體異常相關的惡性腫瘤,也可以預測胎兒的性別有利於性連鎖遺傳病的預防。常用的材料是羊水中脫落細胞與絨毛細胞,一般經組織培養後,製備染色體片,進行核型分析及高分辨顯帶等檢查還可直接作X、Y小體檢查。絨毛細胞也可作短時培養後立即製片觀察。現有資料表明,絨毛細胞染色體分析結果和羊水細胞染色體分析結果不盡一致。歐洲21箇中心1401例檢查的總結指出:a.絨毛的異常染色體率比中孕期羊水細胞高;b.出現的常染色體變異總數比中孕期羊水細胞高3倍,並發現一些不能存活的三體(如14、15、16三體);c.性染色體異常率也3倍於羊水細胞,其中45,X比羊水細胞高10倍;d.夫婦之一有染色體平衡易位者。其絨毛染色體有不平衡易位者也高於羊水細胞;e.絨毛細胞發現染色體異常的復發風險率為4.16%羊水細胞為1.5%。早孕期絨毛細胞與羊水細胞診斷不完全一致至少能提示,在胚胎髮育過程中,自然界的選擇作用不斷淘汰異常細胞而其他的意義尚不清楚。
C.酶學水平:通過對羊水及其中的細胞絨毛細胞或母體血尿的酶學檢查,可以檢出許多先天性酶異常疾病
D.代謝產物水平:檢出特異性代謝產物,可預先診斷某些遺傳性代謝病,如黏多糖病等。
E.基因水平:利用羊水細胞、絨毛細胞或胎兒活檢組織,甚至母體外周血中的胎兒細胞作材料,運用極度敏感和特異性很強的基因診斷技術,檢出遺傳病患兒。
近幾年,隨著體外受精、胚泡體外培養、顯微操作取單細胞人工胚胎移植等技術的發展,出現了一種植入前診斷技術(又稱著床前診斷)。它套用現代分子生物學技術中PCR原位雜交等靈敏度特異度都很高的檢測手段分析體外受精的胚胎或經子宮灌洗所得的胚泡中的單細胞或幾個細胞的基因組成,以明確是否為致病基因攜帶者,並將健康的胚胎移植入母體繼續發育這是產前診斷技術的又一發展,但目前尚不成熟,未能推廣套用
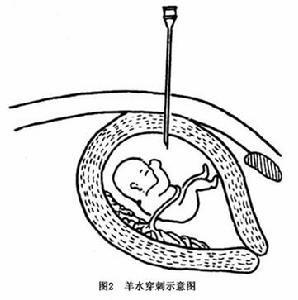
③羊膜穿刺術:受精卵發育第7天形成羊膜腔,生成羊水,羊水與胎兒直接接觸是胎兒發育所需營養的主要供應途徑之一,也是胎尿的排出地,它的成分很能反應胎兒的生長代謝情況。所以羊水及羊水中脫落的胎兒細胞是產前診斷的主要材料,羊膜腔穿刺術的成功套用是標本採集的關鍵。以下介紹羊膜腔穿刺術的技術要點。
A.羊膜腔穿刺的適應證與禁忌證:凡是臨床及其他信息提示需要進行產前診斷的孕婦均為羊膜腔穿刺的適應證。一般認為的禁忌證有:a.妊娠不足12周(子宮太小)或超過24周(細胞培養不易成功);b.適應證不明確;c.先兆流產或稽留流產的孕婦;d.盆腔或宮內有感染者;e.單純因社會習俗要求預測胎兒性別者。
B.羊膜腔穿刺的時間:最好在妊娠16~20周。理由是:
a.此時羊水量大(超過170ml)增長快,抽出20ml羊水不會因宮腔驟小,而引起流產。
b.胎兒與羊水比例較合適,胎兒小,羊水多周圍有較寬的羊水帶,穿刺不易傷及胎兒。
c.羊水細胞中有活力的細胞比例此時最高,易於培養成功。
d.以上皮細胞和成纖維細胞為主的羊水細胞,適合酶和生化分析
C.穿刺的方法:穿刺前需認真作好以下準備工作:
a.核對適應證、妊娠周數、子宮大小、有無併發症。
b.作外周血白細胞、血紅蛋白及血型檢查。
c.檢查穿刺部位的皮膚有無皮炎、感染等不利於穿刺的情況
d.選擇好合適的穿刺部位,可以用B超來幫助定位胎盤及確定是否為單胎,穿刺時也可在B超引導下進行
e.穿刺前孕婦必須排空尿液最合適的穿刺部位在恥骨上3橫指腹中線旁,最好宮底在臍恥之間,或臍下2橫指處,使進針部位剛好在子宮的中心或稍偏下。進針前應仔細觸診。穿刺採用21號長腰穿針頭(帶針芯)。一般步驟:消毒穿刺部位及周圍皮膚,鋪洞巾,局麻,垂直快速進針,刺入皮膚後緩慢進針至7~8cm深(進入宮腔時可有落空感),抽出淺黃色透明液體,即為羊水(圖2)。先抽1~2ml用作生化檢查,沉澱細胞可用作性染色質檢查,另抽15ml放入無菌試管,用於作細胞培養。
D.羊膜腔穿刺中的常見問題:
a.穿刺失敗:一般的失敗率僅為0.5%~1%可能的原因有:子宮太小,或穿刺部位太低,誤穿了膀胱內的尿液;腹壁太厚進針不夠深;穿刺到胎盤附著部位抽出血液後未敢繼續進針再抽。
b.羊水帶血:如開始即抽出血性羊水,提示針尖尚有部分在宮壁宜將針深刺,抽出羊水後換乾淨的針筒再抽即可順利抽出透明羊水。如果羊水抽得很通暢,而始終帶血,則可能針尖刺中胎體或胎盤造成出血而致胎盤前置的孕婦發生的可能性更大
c.對孕婦及胎兒的損傷:一般極少發生,偶見有刺傷孕婦腹壁下動脈,形成大血腫而休克;穿刺在胎盤上,形成胎盤後血腫而致流產;穿刺傷及胎兒皮膚,出生後胎兒身上有點狀凹痕;刺傷胎兒致使一下肢壞死等報導。
d.官腔內感染:因操作時失誤,將細菌帶入官腔,可引起宮內感染和胎兒死亡,故操作時要極端小心,嚴格無菌觀念
e.流產:一般發生率極低。可能由於穿刺針眼羊水外流、出血導致流產
f.Rh血型問題:Rh陰性血型的孕婦,疑有胎兒Rh血型不合時,可在穿刺後給孕婦注射抗D球蛋白,如果胎盤在後壁附著,則可不必。
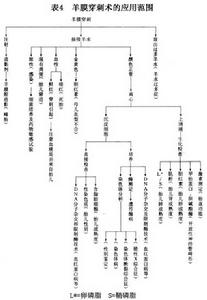
g.羊膜腔穿刺術的套用:表4列出了羊膜腔穿刺術在產前診斷中的套用範圍在此僅對羊水細胞培養的技術細節和羊水的生化檢查作進一步的說明。羊膜穿刺術的套用範圍(表4)。
h.羊水細胞培養的注意事項:羊水細胞培養是為了獲得更多的胎兒細胞以滿足其他檢查的需要羊水細胞大部分為羊膜上皮及胎兒脫落上皮細胞,要培養成功,應注意:有活力的細胞要有一定的比例;培養液的品種和小牛血清要經選擇,HamF10,HamF12培養液成功率較高,小牛血清或牛胚胎提取液含生長素,對促進有活力的細胞生長很重要;促進羊水細胞及早貼壁,一般貼壁時間為5~7天,開始貼壁時多為上皮細胞,換液後成纖維細胞有大片生長;注意影響培養細胞成活的因素培養細胞生長太快或太慢都要考慮母體細胞的污染;離體細胞培養時,會發生染色體的變異。需加以鑑別;其他影響因素:真菌、支原體污染。抗生素的使用,羊水中血細胞的污染及培養的其他條件的影響。
i.羊水的生化檢查:羊水的生化成分的變化可以直接反映胎兒的生長發育情況對其作細緻的生化分析,可以提供很多遺傳病的信息,此類研究現在已越來越多。表5列出了幾種常見的羊水生化指標與其相應的遺傳病。
④產前診斷的意義:產前診斷能在胎兒出生前預先明確是否患有某種遺傳性疾病或先天畸形,通過精確的染色體分析和基因診斷,還可明確是否為某種遺傳變異的攜帶者,為進行臨床疾病防治及遺傳病的各級預防提供最直接的依據。根據臨床資料輔助檢查資料、群體調查資料及產前診斷結果,可以進行綜合分析,及時採取必要措施,如對患病胎兒的選擇性終止妊娠,對可作早期治療的遺傳病(如苯丙酮尿症)進行治療,對有些簡單的先天畸形還可進行宮內手術治療。產前診斷已成為現代優生學的重要基礎方法,它在幫助限制整個人群致病基因的擴散,降低遺傳病發生率,監控出生人群的遺傳素質等工作中起著越來越大的作用。隨著醫療保健條件的改善,產前診斷的適應證不斷擴大,也為醫學遺傳學的研究提供大量的第一手資料。
2.埃萊爾-當洛綜合徵的二三級預防 從遺傳病預防學角度看,遺傳病的治療屬於二級和三級預防的範疇。遺傳病的治療的關鍵是:儘早發現、儘快治療。治療時機的掌握主要有以下幾種:①出生前確診(產前診斷)後,可進行產前治療(宮內治療)或產後立即治療。宮內治療方法有孕婦給藥療法和直接治療胎兒兩類。只要所使用的藥物能通過胎盤,孕婦給藥法就方便、安全易被接受。如孕婦服用生物素、維生素B12腎上腺皮質激素、洋地黃等,可分別治療胎兒的生物素依賴性羧化酶缺乏症維生素B12依賴性代謝性酸中毒、先天性腎上腺皮質增生症和先天性室上性心動過速。對不能通過胎盤的藥物,可直接注入羊膜腔,讓胎兒在吞咽羊水過程中將藥物一併吞食。如甲狀腺素直接注入羊水可治療遺傳性甲狀腺腫。胎兒外科手術治療,現也有成功的報導;②典型症狀出現前予以確診(症狀前診斷)確診後給予儘早治療。例如苯酮尿症患兒可在出生後吃奶72h後用Guthrie血斑濾紙細菌抑制法進行早期確診給予低苯丙氨酸飲食治療,可以防止患兒智力損傷;③各種症狀都出現後才被確診。此時器官組織的損害都已出現,治療方法便不多而且療效欠佳。可採取外科手術(病損器官切除、修補替換等)和內科對症療法來改善症狀。
 埃萊爾-當洛綜合徵
埃萊爾-當洛綜合徵遺傳病治療中總的原則是禁其所忌去其所余,補其所缺,調節代謝平衡防止症狀的出現。
(1)糾正代謝紊亂:這是目前治療遺傳性代謝病的最主要方法隨著對遺傳性代謝病發病機制和中間過程的認識不斷深化,此法的適用範圍也日益擴大。
①飲食控制(禁其所忌):當代謝異常造成機體某些必須物質缺乏時,通過飲食加以補充;而當代謝物質發生貯積時,則限制此代謝物或其前身物質的攝入來維持平衡。苯酮尿症患者低苯丙氨酸飲食就是很好的範例。另外還可通過限制對特定物質的吸收來減少攝入,如苯酮尿症患者服用苯丙氨酸氨基水解酶膠囊,可以將食物中的苯丙氨酸轉化為轉苯丙烯酸,而被消除。
②減少底物(去其所余):因代謝產生有害物質而引起疾病時可以通過降低有害底物和減少其前身物質及代謝衍生物的濃度,去除或減少其毒性作用來控制或改善疾病的症狀。主要方法有:A.螯合或促進排泄;B.血漿置換法和親和結合法;C.改變代謝途徑;D.外科旁路手術;E.代謝抑制。
③產物替代(補其所缺):當重要的酶促反應產物不足而致病時可直接補充相應的必需的終產物。如給垂體性侏儒患者以生長激素給血友病患者以抗血友病蛋白(凝血因子),給遺傳性免疫缺陷病人以相應的免疫球蛋白。
(2)糾正酶活性異常:
①輔酶的補充:有些遺傳病酶活性異常可能累及:
A.一種特異性輔酶或維生素的結合部位。
B.有活性的輔酶轉運或生物合成過程,導致異常。許多輔酶是全酶正常活性所必需的。所以補充輔酶成分也是誘導酶活性增加的一種有效方法,它可以使全酶在細胞內降解速度減慢,提高酶的半壽期,還可降低酶促反應的米氏常數(Km)目前已用此方法治療25種以上的遺傳病。如用鈷胺素(B12)治療多種貧血和甲基丙二酸尿症等。
②酶誘導或反饋抑制:對酶缺陷水平的另一種療法是用藥物來提高殘餘酶活性以改善代謝水平。例如苯巴比妥和有關藥物能明顯刺激滑面內質網的生成並能加速內質網中特異性酶合成包括肝UDP葡萄糖醛酸轉移酶為用苯巴比妥治療Gibert綜合徵和Crigler-Najjar綜合徵提供了理論基礎。
反饋抑制作用是許多代謝調節中的重要形式,針對因某種酶缺陷引起的底物或其前體堆積,可以通過其他旁路代謝的反饋抑制作用來提高酶活性,減少堆積的底物,反饋抑制已作為治療急性卟啉症的一種方法。
③同種移植:通過向遺傳病個體植入同種含正常基因的細胞,組織或器官,以期在受體內產生相應的有活性的酶及其他基因產物,達到治療目的。移植物在受體內可能通過兩種機制發揮作用:
A.產生活性酶,在原位代謝除去原來的貯積底物。
B.釋放活性酶、輔酶或免疫活性因子入血分布到全身其他組織中發揮作用。至今已進行過此類同種移植的組織器官有:腎、肝腎上腺、骨髓、胸腺脾、胰等有的已取得明顯療效。
④酶替代療法:直接給酶缺陷患者提供相應的正常的酶。隨著酶學技術和細胞工程基因工程技術的發展,已經可以提供足量的、高純度的酶製劑。這種酶製劑必須具有半衰期長、抗原性低、導向性好等特性。為此常採用的方法是:
A.採用微囊、脂質體、紅細胞影泡等載體來包裝酶製劑,以減小免疫原性,延長半衰期。
B.套用受體介導分子識別法來提高導向性
C.對一些溶酶體貯積病,因其沉積物可以彌散入血,並保持動態平衡,則可用“平衡一去除”法來治療。
(3)基因治療:基因治療是指運用基因轉移技術直接將遺傳物質導入生殖細胞或體細胞以起到對遺傳病及其他疾病的治療作用的新型治療方法。對遺傳病進行基因治療可望從根本上糾正遺傳病的表型異常。
①基因治療的基本策略:近10餘年來,基因治療研究蒸蒸日上,提出了許多新思路、新構想,目前主要的策略有:
A.基因的原位修正(correction)和原位替代(replacement):這一策略的目的就是要將突變的基因在原位修復,而不影響其周圍其他基因的結構和功能其中原位修正針對基因的點突變或小範圍變異,擬通過特定方法對其定點修復。而原位替代,就想把有較大範圍變異的基因去除而換之以正常的基因。這一策略是最理想、最直接的對遺傳變異進行根治的方法,目前研究很多的哺乳動物細胞內定點整合(同源重組),給這種策略提供了理論和實驗依據,但至今未能真正用於人體試驗。
B.基因增強(gene augmentation或gene complementation):在不改變缺陷基因本身的前提下,將外源有功能的基因轉移到疾病細胞或個體基因組內,使其表達以補償有病基因失去的功能此策略是目前研究最多也是最成熟的方法
C.將反義基因或其他對抗異常基因表達產物的基因導入細胞內,起到抑制作用,或稱基因抑制療法(gene inhibition therapy)或細胞內免疫(intercellular immunity)。
②基因治療的技術要點在基因治療的諸多策略中研究最多、最成熟並套用於臨床試驗的是基因增強的策略。整個研究過程通常包括臨床前研究和臨床研究(表6)。
A.疾病的選擇:目前基因治療首選的是單基因缺陷性疾病。選擇的基本條件常包括:
a.遺傳基礎比較明確,目的基因能在體外克隆
b.基因表達不需精細調節,而且經常開放,產物生理水平不高者更佳。
c.具有一定發病率,危害較大,尚缺乏其他有效治療措施者
我國是開展基因治療研究較早的國家之一復旦大學薛京倫等就是根據這些條件,選擇血友病作為研究對象,已取得了很好的結果,達到了世界先進水平。當然這些條件是限於現有的研究水平才提出的。
B.靶細胞的選擇:基因治療的靶細胞可分為兩大類:生殖細胞和體細胞。由此引出了生殖細胞基因治療和體細胞基因治療的分類。如果能對生殖細胞或早期胚胎細胞進行基因修復或替換、使基因缺陷得到校正,使遺傳病不但能在當代得到治療,還能將新基因傳給下一代,也為人群減少1個有害基因,是理想的遺傳病根治手段。但是,由於現代生物技術、理論的限制,以及生殖細胞基因操作涉及人類社會的倫理、道德和法律等多種因素,在相當長的一段時間內只能進行動物試驗。1985年美國政府就已規定,把基因治療的人體試驗限制在體細胞。已經被用於作為靶細胞的有:造血幹細胞、肝細胞、成纖維細胞內皮細胞、淋巴細胞等。
C.基因轉移的載體和轉移方法:構建合適的轉移並表達的載體和選擇高效的基因轉移方法是基因治療的關鍵,常用的載體有:逆病毒載體質粒載體和腺病毒載體腺相關病毒載體,另外還有脂質體載體。常用的基因轉移方法有4大類型:
a.化學法:主要是磷酸鈣沉澱法。
b.物理法:常用電導和顯微注射法。
c.膜融合法:以脂質體包裹法較好。
d.病毒法:主要指反轉錄病毒和腺病毒介導的基因轉移。
③基因治療的前景:基因治療概念的提出已有幾十年的歷史,只到了近10年,隨著現代分子生物學技術(特別是DNA重組技術)的發展,這一概念才得到有力的理論基礎和技術方法的支持並得以付諸實施。1990年,2名腺苷脫氨酶(ADA)缺陷引起嚴重免疫缺陷的患者接受基因治療獲得成功,這標誌著基因治療的研究進入了一個新的階段從此世界各國的生物醫學家在各國政府部門及社會各種力量的大力支持下,全面展開了基因治療的研究。由原來針對單一的遺傳病發展到腫瘤、傳染病等多種疾病,提出了基因調控療法、基因抑制療法等新概念、新途徑。到1994年上半年已有100多個臨床試驗方案獲準實施有的已取得很好的效果。當然,基因治療發展的歷史還不長,要廣泛套用於臨床還需大量的研究探索,尤其是以下幾方面的問題:
A.對更多遺傳病的分子基礎及基因表達調控機制更深入的了解,這是基因治療的基礎。
B.構建更有效和安全地表達並轉移的載體。
C.更簡便有效的基因轉移方法的建立
D.定點整合、原位修復系統等技術的完善。
E.更多更接近實際的動物模型(尤其是轉基因動物模型)的建立,這是基因治療臨床前試驗的必由之路。
F.體細胞基因治療、生殖細胞基因治療等的倫理學及相關的科技管理立法等方面的探討。
G.還需充分考慮基因治療可能存在的危害性,如插入突變導致的嚴重後果、缺陷病毒載體經重組後恢復感染性的危害及外源基因轉入體內的其他潛在危害等。總之,我們認為基因治療作為一種惟一從基因缺陷本身入手,可望徹底治療遺傳病新型治療途徑有非常吸引人的前途,但仍需從基礎理論、技術方法及倫理道德等多方面進行深入廣泛的研究探索,才能適應現代醫學模式被人們所接受真正成為人類防病治病的有效手段。
