
疾病概述
 先天性非溶血性黃疸
先天性非溶血性黃疸症狀體徵
 先天性非溶血性黃疸
先天性非溶血性黃疸新生兒生理性黃疸具有以下特點:①黃疸出現時間:在生後2—3天出現。②黃疽程度:屬輕度到中度黃染,呈淺杏黃色或黃紅色帶有光澤,進展緩慢。⑧黃疸尖峰時間:在生後4—5天。④血清總膽紅素值:足月兒一般不超過205.2umol/l早產兒一般不超過256.5umol/L。⑤黃疸消退時間:一般在生後7—10天左右,足月兒最長不超過2周;早產兒不超過4周。⑥伴隨症狀:除黃疽外,無貧血或肝脾腫大等症狀,嬰兒一般情況良好。早產兒的黃疸出現時間可能遲一些,程度可重一點,消退時間也可晚一些。新生兒生理性黃疸的膽紅素值可因民族、地區、圍產期產婦的情況以及新生兒個體情況而不同。生理性黃疸一般不須治療,只要提早餵養,供給足夠的熱量及液量,保持室內空氣流通及光線充足,即可使黃疸程度減輕及消退加快。
診斷標準凡具下列其中之一者,應考慮為病理性黃疸:①黃疸在生後24小時內出現。②總膽紅素一般足月兒>205.2umol/l,早產兒>256.5umol/L。⑧黃疸進展迅速,總膽紅素每24小時升高的速度超過86umol/L。④結合膽紅素>26umol/L。⑤黃疸持續時間延長(足月兒超過2周;早產兒超過4周),或生理性黃疸消退後又復出現,或進行性加重病人一般情況尚可,多無明顯自覺症狀;部分病人可伴有易疲勞、肝區不適、消化不良等。有時Gilbert綜合徵患者也可伴有輕度溶血性貧血。除偶見顯性黃疸外,無異常體徵,肝脾常不腫大。根據血清膽紅素的濃度不同,可將本綜合徵分為輕型和重型。輕型較重型多見,血清膽紅素低於85.5μmol/L;重型的血清膽紅素大於85.5μmol/L,常在新生兒期即出現黃疸。
病因及病理
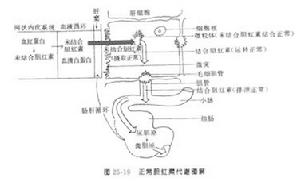
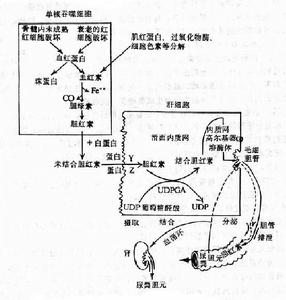
目前多數人認為由於遺傳性或獲得性的肝細胞中微粒體器中膽紅素葡萄糖醛酸轉移酶活力不足影響非結合膽紅素在肝細胞內結合反應的正常進行,以致使肝細胞對膽紅素的攝取也受到障礙,因而造成肝細胞對非結合型膽紅素的攝取和結合功能的雙缺陷。病理生理
在所有病人肝穿刺活體組織標本中,證實肝臟的膽紅素葡萄糖醛酸轉移酶的活力值明顯減低,提示肝臟從血漿中清除間接膽紅素的能力降低,但血漿非結合膽紅素的濃度與該酶活力降低的程度無明顯關係。這可能由於部分Gilbert綜合徵病人也同時存在著緩和的代償性溶血情況所致;從膽紅素轉運動力學研究提示,非結合高膽紅素血症的原因不是由於生成過多,而是由於轉運缺陷。另一方面通過部分病人伴有BSP轉運異常,也提示本綜合徵有部分病人具有轉運功能缺陷,由於游離膽紅素進入肝細胞後,被肝細胞漿內的兩種低分子可溶性“受體蛋白”(Y、Z蛋白接受)帶到滑面內質網,在酶的作用下進行結合,若Y、Z蛋白量不足或接受功能差時,則造成運輸障礙也會影響肝細胞對非結合膽紅素的攝取與結合,根據血清膽紅素的濃度不同,可將本綜合徵分為兩型,其發病機制可能有所不同。
1.輕型較重型多見,血清膽紅素低於85.5μmol/L,糞內尿膽原正常。其發病機制可能為肝細胞攝取及轉運非結合膽紅素的過程有缺陷。如肝細胞漿內可溶性蛋白受體不足或其接受功能不良,造成肝細胞內非結合型膽紅素的轉運障礙,而影響了肝細胞對非結合型膽紅素的攝取不良。但也可能有一部分輕症病人與重症病人的發病機制相同,是屬於同一類型的,即由於葡萄糖醛酸轉移酶活力減低不明顯所致,但由於缺乏敏感檢測技術,而與測不出極輕度酶活力的降低有關。
2.重型血清膽紅素大於85.5μmol/L(5mg/dl),常在新生兒期即出現黃疸。由於同時伴有肝細胞中微粒體內葡萄糖醛酸轉移酶活力不足,而致肝細胞結合功能不良,造成非結合膽紅素增高血症的黃疸。
診斷檢查
 先天性非溶血性黃疸
先天性非溶血性黃疸1.慢性間歇性或波動性輕度黃疸,有發作誘因,可有家族史,一般狀況良好,無明顯症狀。
2.體格檢查除輕度黃疸外,無其他異常體徵,肝脾多不大。
3.一般肝功能(ALT、AST、AKP、膽汁酸)正常,僅有血漿非結合膽紅素增高水平的波動性升高。
4.無溶血性、肝細胞性、阻塞性黃疸證據。
5.肝組織病理學檢查正常。
如在12~18個月內經2~3次隨訪,無其他實驗室異常發現,即可診斷為Gilbert綜合徵。檢測UGT1啟動子內TATAA序列或基因有無突變有助於診斷。
實驗室檢查:大多數病例的黃疸輕微,血清總膽紅素在22.1~51.3μmol/L,少數至85~102μmol/L或更高,主要為血中非結合膽紅素升高。血清膽酸正常,其他肝功能試驗正常(如ALT、AST和γ-GT)。無溶血證據,紅細胞脆性試驗正常。尿膽紅素陰性,糞中尿膽原量正常,尿中尿膽原量不增加。
其他輔助檢查:
1.膽囊顯影良好,膽囊造影可無異常。
2.苯巴比妥試驗苯巴比妥能夠誘導肝臟微粒體葡萄糖醛酸轉移酶的活性,促進非結合膽紅素與葡萄糖醛酸結合,降低血漿非結合膽紅素的濃度。口服苯巴比妥2周,3次/d,每次60mg;服完藥物後測定血漿膽紅素的濃度,多數病人黃疸改善,血清間接膽紅素明顯下降,甚至可達正常;如系UGT1的完全缺如所引起的黃疸則無效。
3.低熱量飲食試驗2~3天內每天給予1674kJ(400kcal)飲食,若血漿間接膽紅素值增加大於100%,或增加25.65μmol/L,有診斷意義。恢復正常飲食後12~24h,降至基礎水平。低熱量飲食試驗對本病的敏感性約80%,特異性幾達100%。飢餓引起Gilbert綜合徵患者血清膽紅素升高機制可能是多因素的,與飢餓引起的下列改變有關:肝內膽紅素配體和Z蛋白含量降低;血紅素分解代謝增加;脂肪組織內脂解,游離脂肪酸增加,引起膽紅素游離和釋放入循環;腸蠕動減弱,膽紅素腸肝循環增加。
4.給Gilbert綜合徵病人示蹤劑量的放射性核素標記的間接膽紅素,並測定24h後在血漿中存留的百分數,Gilbert綜合徵病人的數值比正常人增高。
5.肝活檢查無明顯改變,偶可見少量脂肪性變,偶在終末性肝血管周圍有脂褐素樣色素沉著。肝穿刺取活體組織做膽紅素葡萄糖醛酸轉移酶活力測定,其活力較正常人明顯減低。電子顯微鏡檢查,可見到肝細胞內的粗面內質網及其上的蛋白微粒均顯著減少,滑面內質網則增加肥大。
鑑別診斷
需要與脂肪肝、酒精中毒、慢性膽囊炎、肝硬化及病毒性肝炎等引起的慢性非結合性高膽紅素血症相鑑別;還需要與慢性溶血性黃疸鑑別,後者除間接膽紅素增高外,尚有貧血、網織紅細胞增高,尿中尿膽原亦有所增高。部分血清病原學陰性病毒性肝炎被誤診為Gilbert綜合徵的原因有:病史不詳、片面重視治療試驗及飢餓試驗、對肝穿刺活組織檢查缺乏重視。
治療方案
 先天性非溶血性黃疸
先天性非溶血性黃疸併發症
可有輕度溶血性貧血。
預後及預防
預後:Gilbert綜合徵系一良性疾病,良性經過,預後良好。
預防:目前尚無相關資料。流行病學(查看內容)先天性病人家族中約有25%~50%的人有此病,為常染色體顯性遺傳病。患者主要為兒童、青少年,男女比例為2∶1~7∶1。
