疾病描述
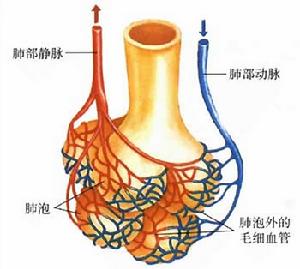 毛細血管
毛細血管症狀體徵
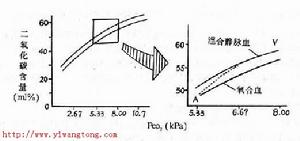 低氧血症
低氧血症1.腎臟改變MPA主要特徵為100%腎臟受累,大部分患者表現RPGN、少尿或無尿、血尿,且1/3呈肉眼血尿、蛋白尿、嚴重者可見腎病綜合徵範圍蛋白尿,嚴重高血壓並不多見,腎功能可呈進行性減退。
2.肺受累12%~29%本病患者伴有肺出血,也是發病和死亡的重要因素之一。咯血為常見肺部受累表現,輕則痰中帶血絲,重則大量咯血。多數病例在入院1月內出現,但亦可長期存在,伴呼吸困難和貧血,肺出血可導致嚴重低氧血症。常見影像學特徵為肺泡陰影而無肺水腫或感染。一氧化碳轉換係數升高(≥30%)亦提示肺出血,可依靠支氣管肺泡灌洗法確診。部分小血管性肺血管炎患者臨床影像學功能檢查符合間質性病變過程,類似特發性肺纖維變性。
3.其他臨床表現與PAN相類似,65%~72%患者有骨骼肌受累(肌痛、關節痛、關節炎);44%~58%有皮膚改變(紫癜、片狀出血);胃腸道症狀有腹痛(32%~58%)和消化道出血(29%);僅14%~36%患者有外周神經病變,較PAN少見;眼、耳、鼻、喉等處病變較PAN多見,個別患者有口腔潰瘍。
病理病因
 白細胞
白細胞病理生理:20世紀80年代以來,抗中性粒細胞胞質抗體(ANCA)對血管的致病作用日益引起高度重視。ANCA在主要表現為壞死性腎小球腎炎的MPA中陽性率明顯升高。致病抗原的主要成分是白細胞髓過氧化酶(MPO)或蛋白酶3(PR3),產生相應的特異性白細胞胞質抗體,造成血管壁損傷。ANCA學說引起國際廣泛興趣,目前正在積極探索研究中。引起血管炎的致病因子可能是複合而並非單一的。近來,ANCA被認為是血管炎內皮損傷發展的重要病因,尤其在WG和MPA中。體外實驗示ANCA可刺激中性粒細胞黏附於內皮細胞並誘導TNF-α(腫瘤壞死因子-α)致敏的白細胞溶解培養中的內皮細胞。由於ANCA的靶抗原MPO和PR3僅存在於噬天青顆粒中,故ANCA-PMN(多形核中性粒細胞)激活機制尚未闡明。曾從致敏PMN胞質膜上測及PR3,而後在體外和離體研究中示TNF-α及IL-8的協同作用於PR3,使其由顆粒內移至中性粒細胞膜上。
細胞因子誘發黏附分子的表達(LFA-1,ICAM-1和ELAM-1)致PMN和內皮細胞緊密接觸,細胞因子致敏的中性粒細胞、內皮細胞和血循環中ANCA的共存使ANCA引發後續級聯反應而發生血管炎。儘管實驗與臨床均有所發現,但PR3-ANCA和MPO-ANCA在血管炎發病中的具體作用仍不詳。
 腎活檢
腎活檢診斷檢查
 免疫螢光
免疫螢光實驗室檢查:本病尚無特異性檢查。
1.血液檢查主要為血沉增快,血小板和白細胞計數增多,少數患者存在嗜酸性粒細胞增多症,血紅蛋白降低呈正細胞性貧血;血漿白蛋白水平下降。幾乎所有患者均為B肝表面抗原陰性,C-反應蛋白增高,α2-球蛋白水平上升,總補體、C3、C4水平正常或部分升高,39%~50%患者RF陽性,較抗核抗體(21%~33%)多見。
2.腎功能檢查所有患者均有腎功能受累,血肌酐>120μmol/L。Jerra組中15%血肌酐水平正常,Hammersmith組中血肌酐平均值為574μmol/L(波動於147~1405μmol/L)。常伴鏡下血尿,超過90%的患者有蛋白尿,多>3g/24h。
3.ANCA檢測用間接免疫螢光(IFT)、酶聯免疫吸附法(ELISA)等方法檢測。IFT因抗核抗體而常有假陽性,故IFT結果一定要結合ELISA測靶抗原抗體才有意義。IFT結合ElLSA法診斷小血管炎特異性高達90%。IFT法檢查有兩種圖形:胞漿型C-ANCA,粒細胞均勻著色,核周型P-ANCA,著色集中在分葉核的核周邊。
本病IFT主要表現為核周型P-ANCA,ELISA測靶抗原為髓過氧化物酶(MPO)。活動期陽性率50%~75%以上。國內張少凌測19例MPA,抗MPO陽性6例,全部並腎損害,5例並肺損害。提示抗MPO抗體多見肺、腎病變,活動期前1月即可升高,緩解期下降。
其他輔助檢查:
1.組織活檢小動靜脈病變同經典型PAN。鑑別在於腎病理檢查,腎臟活檢可見局灶節段壞死性腎小球腎炎(FSNG)並新月體形成,免疫螢光多數陰性。80%急進性腎炎Ⅲ型(無免疫複合物無螢光反應)為微血管炎引起。
2.血管造影無微動脈瘤及狹窄。
鑑別診斷
 鼻旁竇
鼻旁竇2.古德帕斯丘(Goodpasture)綜合徵症狀與MPA相似均有肺出血和急進性腎炎,但血中可找到抗基底膜抗體,腎病理免疫螢光有特徵性腎小球基底膜線性IgG、C3沉著。
3.經典型結節性多動脈炎經典型多侵犯中等動脈及其分支處,而後者累及小動脈及小靜脈,特點是中小動脈壞死性、非肉芽腫性血管炎,腎小球腎炎是經典型結節性多動脈炎與微型多發性動脈炎兩者的主要鑑別點,如中等動脈及小動脈同時受累,即稱為結節性動脈炎重疊綜合徵。兩病的臨床表現及預後不同。
治療方案
 類固醇激素
類固醇激素1.皮質類固醇激素和環磷醯胺(CTX)治療初始,宜大劑量套用皮質類固醇激素和環磷醯胺(CTX)。重症患者的首次治療多用甲潑尼龍衝擊療法,劑量為15mg/(kg?d),1h內靜脈推注,連用1~3天,本療法起效快,相對安全,尤其適宜於多器官受累的危重患者。衝擊治療3天后改為潑尼松1.0mg/(kg?d)或等效甲潑尼龍,晨起一次頓服。通常1個月內臨床症狀改善。血沉恢復正常時激素可減量維持,1年後激素可緩慢停藥。當激素與環磷醯胺(CTX)合用時,激素減量宜加快以免並發感染。常規定義小劑量環磷醯胺(CTX)為2mg/(kg?d)或療程少於1年。傳統治療中常與皮質類固醇激素聯合套用,雖然對血管炎治療有效,但其治療量/中毒量比值低。環磷醯胺(CTX)主要不良反應包括:出血性膀胱炎,膀胱纖維變性,骨髓抑制,卵巢失功能和腫瘤(膀胱癌和惡性血液系統疾病),嚴重感染是血管炎患者主要致死因素,尤其在大劑量套用激素和免疫抑制劑時。
為減少每天口服環磷醯胺(CTX)所致死亡,近來發展至大劑量間歇服用藥物治療,劑量、總量和頻度均根據患者病情、腎功能、血液指標及對治療敏感性調整。據法國PAN協作研究組資料示,環磷醯胺(CTX)衝擊劑量0.6g/m2,每月1次,持續1年。對於腎功能衰竭患者大劑量靜脈推注環磷醯胺(CTX)尤為危險,因而提示應慎重減量。建議在衝擊時充分水化,並使用泌尿道保護劑2-巰乙磺酸鈉(美斯納)0.4g,3次/d靜脈推注。該療法環磷醯胺(CTX)累積量小,患者僅短期內有毒副作用。
2.血漿置換(PE)治療該法可改變機體的自身抗體與抗自身抗體之間的平衡,從而有利於抑制致病抗體。但目前尚無理由支持在MPA確診時即行血漿置換,PE可適用於腎衰患者(肌酐>500μmol/L)或透析依賴者和肺出血患者。Pusey報告了一組前瞻對照研究,在血管炎引起局灶性壞死性腎炎而致急性腎衰依賴透析的患者中,PE治療組10/11例恢復了腎功能,而藥物組僅3/8例恢復了腎功能,故認為對於依賴透析的急性腎衰或伴有嚴重肺出血的血管炎患者應首先予PE治療。
3.支持治療暴發性MPA常表現為肺-腎功能衰竭,因肺泡氣體交換下降致全身氧轉運障礙,加之失血導致貧血,大量肺泡出血需立即補充液體並給予循環呼吸系統。儘管肺出血在治療起始後迅速緩解,但其致死率高,預後差。對於那些具潛在死亡因素的患者而言,支持療法亦是治療的重要組成部分。因嚴重胃腸道受累致體重快速急劇下降者需予以腸外營養。治療初始免疫抑制程度最甚,故應對機會性感染如卡氏肺囊蟲性肺炎預防性治療,可因人而異。對多神經炎的治療以控制疼痛、預防壓迫性潰瘍和理療為主。
4.腎功能惡化時需行血液透析。
5.免疫球蛋白靜脈注射免疫球蛋白儘管在PAN和MPA治療過程中有重要作用,仍需進一步探討免疫抑制劑的最佳套用指征以減少治療毒性,並評價新的免疫調節藥物如靜注免疫球蛋白或單克隆抗體結合物。
靜脈注射免疫球蛋白最有用之處是在成功治療川崎病上,可預防冠狀動脈動脈瘤的發展,顯著優點為無嚴重不良反應,可用於治療WG和MPA。Jayne報導12例患者(其中7例WG、4例MPA),多數患者在常規治療下疾病仍呈活動性,經靜脈注射免疫球蛋白治療後全部獲得改善,並減少了對免疫抑制劑的需求量,平均ANCA水平下降50%。該項研究示靜脈注射免疫球蛋白在WG和MPA兩病的治療中可誘導並維持緩解。最近靜脈注射免疫球蛋白被套用於15例對傳統療法反應差的ANCA相關性血管炎患者,並未得到驚人結果,其中40%患者有所改善,但並無完全緩解者。
以T細胞為靶抗原的單克隆抗體代替免疫抑制劑,亦套用於系統性血管炎的治療中。Lockwood等人在2例免疫抑制劑治療無效的MPA患者中聯合套用CAMPATH-lH(一種能識別淋巴細胞CDw52抗原的抗淋巴細胞單克隆抗體)和抗CD4單克隆抗體,均取得持久的療效。因例數較少此療法尚有待進一步驗證。
預後預防
 抗生素
抗生素認為PAN為一自限性疾病,一旦誘導緩解即不再復發,而MPA復發常見。復發類型未必與原發病表現相似,可累及新的器官。一般而言,復發表現較原發病輕,且多有皮疹和關節痛。MPA的高復發率證實了延長激素或免疫抑制劑治療的合理性。此類情況下硫唑嘌呤對維持MPA的緩解狀態有意義,中斷治療後MPA患者的復發率高(33%),減量治療期間亦可有復發。但CTX並不能防止復發,在復發組和非復發組的CTX療程和總量無明顯差異。有報導慢性活動性病變的預後較長期穩定非活動期血管炎更差。
預防:本病病因不明,目前無有效預防措施。一般認為好發人群日常生活中注意自我防護,避免不健康的生活方式,並做到合理套用抗生素、預防發生過敏反應。尤其對高敏體質人群,更應注意避免各種致敏因素,可有望預防引發本病。
流行病學:年發病率2.4/100萬,男性較女性更易累及,男女比例為1.8∶1。平均發病年齡在50歲左右。
併發症:19%~33%患者合併高血壓,12%~29%本病患者伴有肺出血,重則大量咯血,如長期存在可並發伴呼吸困難和貧血。
