
概述
 遺傳性感覺神經病
遺傳性感覺神經病遺傳性感覺神經病歸類於遺傳性周圍神經病,這是一組由遺傳因素引起的以周圍神經受損為主的疾病。遺傳性感覺神經病多呈顯性遺傳,典型表現為皮膚因感覺缺失而致四肢末端反覆發作性無痛性潰瘍,深感覺缺失所致步態不穩。運動障礙不明顯。
本病多為常染色體顯性遺傳,多為近親結婚所致。發病機制:發病機制尚不清楚,主要病理變化為脊髓後根和脊神經節萎縮變細,以腰髓段脊神經為明顯。
鏡檢發現神經節中的神經細胞變性消失,從神經節發出的神經纖維髓鞘和軸突變性。這種改變主要發生在細纖維。脊髓後索萎縮部分脫髓鞘間質結構組織及施萬細胞增殖,脊髓的前角細胞及脊神經前根多正常。骨骼肌一般無明顯變化。耳蝸神經和前庭神經節也可出現萎縮神經細胞變性減少。部分病人可有趾骨破壞,脛腓骨骨質增生。
病因
 神經節
神經節本病多為常染色體顯性遺傳,多為近親結婚所致。
發病機制:
發病機制尚不清楚,主要病理變化為脊髓後根和脊神經節萎縮變細,以腰髓段脊神經為明顯。
鏡檢發現神經節中的神經細胞變性消失,從神經節發出的神經纖維髓鞘和軸突變性。
這種改變主要發生在細纖維。
脊髓後索萎縮部分脫髓鞘間質結構組織及施萬細胞增殖,脊髓的前角細胞及脊神經前根多正常。骨骼肌一般無明顯變化。耳蝸神經和前庭神經節也可出現萎縮神經細胞變性減少。
部分病人可有趾骨破壞,脛腓骨骨質增生。
症狀
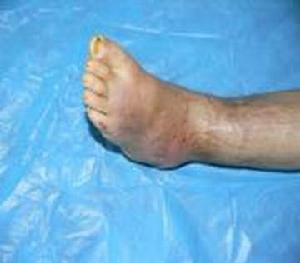 脛骨骨髓炎
脛骨骨髓炎l臨床表現:
遺傳性感覺神經病分類屬於遺傳性周圍神經病,自幼兒期至成年期(30~40歲)均可發病。初發症狀常為始自足部的潰瘍。潰瘍為無痛性和頑固性,易發生在持重足處及足底部。可反覆發作,若為深部穿透性潰瘍則可使受累局部骨骼裸露,趾骨脛骨和肋骨可發生骨膜炎與骨髓炎。由此可致趾間關節,踝關節疼痛、活動受限。部分病人可有足趾變形縮短,甚至可出現足趾脫落。足背動脈搏動正常,感覺障礙的範圍較廣,一般下肢較上肢重常有麻木、蟻走感。有時可表現脛前部、腹部的疼痛。神經系統檢查可見感覺障礙的分布呈手套、襪子型。以痛溫覺受損為重。位置覺震動覺受損相對較輕,有時可表現為淺深感覺分離性感覺障礙。一般無明顯運動障礙,但可有踝反射肱橈反射減弱或消失,自主神經障礙一般不明顯,有時可出現手掌與足部泌汗異常腦神經受累,常表現為耳聾或視神經萎縮,其他腦神經一般不受影響。
根據其遺傳形式發病年齡與感覺障礙的勱不同點,本病可分為4種類型:
Ⅰ型:為常染色體顯性遺傳病,兒童期至成年期均可發病,主要表現為四肢遠端的感覺缺失,以雙下肢為著,痛覺和溫度覺障礙較觸壓覺為重,無感覺性共濟失調,有些病人可表現為撕裂性疼痛。受累肢體汗液減少或無汗。
Ⅱ型:為常染色體隱性遺傳性疾病,於嬰幼兒期發病,故又稱為先天性感覺神經病。主要表現為肢體皮膚各種感覺的消失,以四肢遠端為著。常有甲溝炎手指和趾部潰瘍等。多數肢體亦有出汗減少或無汗。
Ⅲ型:為嬰兒和兒童中的一種隱性遺傳病臨床,特點為嬰兒期起病有餵養不良史,常有嘔吐和肺部感染髮生。患兒對痛覺不敏感,自主神經功能紊亂,包括淚液分泌缺陷,體溫控制不良出汗過多,高血壓和直立性低血壓等。部分患兒表現為角膜反射遲鈍,舌面蕈狀乳頭消失。
Ⅳ型:為一種隱性遺傳性感覺神經病,據認為,本病是由於神經嵴分化異常所致。主要表現為對疼痛不敏感,汗腺分泌減少或無汗。伴有智力遲鈍是本型的特點。
併發症:隨病情發展,可以出現多樣的症狀體徵。
診斷
 遺傳和基因分析
遺傳和基因分析根據:
本病感覺缺失,四肢遠端尤其是下肢反覆發作的頑固性穿通潰瘍,趾骨脫落,缺如,足變形,運動障礙較輕,有家族遺傳史,診斷不難。
鑑別診斷:本病應與下列疾病相鑑別:
1,脊髓空洞症多為一側或兩側上肢的分離性感覺障礙,呈馬褂或半馬褂樣分布,表現痛溫覺障礙而觸覺及深感覺正常。受累肢體可有肌肉萎縮皮膚乾燥無汗,指甲變脆,夏科關節等自主神經功能障礙等表現與本病的感覺障礙不同,易於鑑別。
2,麻風病有典型的皮膚損害,周圍神經(尺、橈、耳大神經等)肥大,受損皮膚的痛覺、溫度覺受累,嚴重損害區不規則,查麻風桿菌陽性可與本病區別。
3,澱粉樣變性具有腹瀉,便秘等消化系統症狀;陽痿泌汗異常與直立性低血壓等自主神經症狀;足尖與下肢異常,感覺痛溫覺障礙等特點。直腸黏膜及周圍神經活檢在組織內有澱粉樣沉積物可與本病區別。
檢查
 遺傳性感覺神經病
遺傳性感覺神經病實驗室檢查:
血常規、生化、腦脊液檢查均正常。
其它輔助檢查:
1,肌電圖檢查多數正常,部分病人表現為感覺神經傳導速度變慢,以下肢較明顯。
2,X線檢查可發現趾骨,跖骨破壞,骨質溶解或增生,骨膜增厚。
3,肌肉活檢正常,或呈失神經性改變,橫紋消失,有散在變性的肌纖維,周圍神經活檢,發現細小無髓纖維幾乎完全消失,有髓纖維脫髓鞘。
治療
本病無特殊療法,對周圍神經損害可用維生素B1、維生素B12、三磷腺苷(ATP)等治療。對本病所致的潰瘍行局部用藥治療。也可配合針灸,理療等治療。
基因診斷
 基因診斷
基因診斷分子遺傳學的發展極大增加了人們對遺傳性周圍神經病發病機制的認識,已對臨床實踐和遺傳諮詢產生重要影響。
目前在常染色體顯性遺傳型和X連鎖顯性遺傳型CMT1、DSS及HNPP均可用突變分析。通過雙列RELPs(限制性片段長度多態性)劑量差異分析或用STR標誌物檢測三倍等位基因均可以闡明染色體17p11.2區域的CMT-1A串聯複製和HNPP缺失。脈衝場凝膠電泳(PFGE)分析是百分之百可靠的診斷方法,其可直接檢測新的連線片段。也可用克隆的CMT-1A-REPs片段做探針用Southen雜交檢測連線片段和或劑量差異。從細胞遺傳學角度開發的中期核螢光原位雜交(FISH)可檢測染色體17p11.2區域的CMT-1A複製和HNPP缺失。也有人採用單鏈構型多態性(SSCP)和DNA直接測序檢測周圍髓鞘蛋白基因PMP22、MPZ及Cx32的點突變。目前這些遺傳性疾病的DNA診斷已無須家庭其他成員的配合,可為每一位有新突變的患者進行遺傳諮詢。
CMT2、HMN、HSN及HNA基因定位結果顯示這類疾病是遺傳異質性的,目前尚不能進行直接的基因診斷。在一些大家系獲得結論性的連鎖信息之後,疾病相關表型的存在或缺失才可作為診斷工具。
預防
預後:由於本病病程進展緩慢,大多數患者可存活數十年,對症治療可提高患者生活質量。
預防:進行遺傳諮詢預防措施,包括避免近親結婚、攜帶者基因檢測及產前診斷和選擇性人工流產等防止患兒出生。
病例
7歲女孩,第一胎足月產,分娩時有臍帶繞頸史。生後6個月會坐,1歲才能爬,一歲半會翻身,至今不能獨自站立,扶持下行走困難。神經系統檢查:四肢肌力Ⅳ度,雙下肢肌肉萎縮,四肢腱反射普遍消失,雙下肢遠端深淺感覺均減退,弓形足。肌電圖顯示肌肉失神經改變,聽覺及視覺誘發電位均延遲。腓腸神經電鏡檢查顯示:(1)神經纖維數目明顯減少,有大量膠元纖維存在;(2)少數神經纖維軸索周圍有數層同心圓板層髓鞘形成;(3)多數軸索因許旺細胞系膜的卷繞和延伸障礙而形成許多典型的蔥皮球,提示髓鞘形成不良。
Gabreels-Festen及Gabreels提出了一種非常罕見的HMSNⅢ型的亞型——伴有典型蔥皮球結構的Ⅲ型。該亞型文獻上僅報導了8~9個病例,均無家族史,故其遺傳類型不清。他們除了腓腸神經活檢均顯示典型的大蔥皮球結構(由同心圓排列的許旺細胞薄板所形成)外,絕大多數病例的運動神經傳導速度延緩,甚至測不出。運動發育延遲,行走困難,甚至不能行走。腱反射消失,感覺障礙。多數病人呈現弓形足,脊柱彎曲和感覺性共濟失調。腦脊液的蛋白含量顯著增高。有1例5歲時死亡。本例考慮屬於此亞型。
