病因
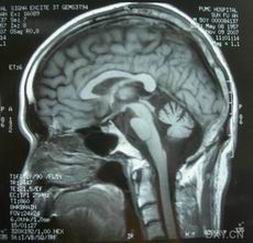 多系統萎縮
多系統萎縮發病機制:
1,SND和散髮型OPCA、SDS均歸類在MSA中,屬MSA不同的3個綜合徵。
2,病理改變:病理改變主要表現在與軀體特定區域有關的黑質緻密部及豆狀核,黑質神經元廣泛喪失,無Lewy小體,在其餘的細胞中沒有神經元纖維纏結,豆狀核變性最明顯,雙側殼核對稱性嚴重萎縮減小,呈灰色,而尾狀核不太明顯繼發的蒼白球萎縮(主要為紋狀體蒼白球纖維喪失)。
鏡下所見:基底神經節顯示殼核疏鬆,神經元幾乎全部喪失,還有大量廣泛散在的細胞外棕色粗顆粒這些顆粒,為鐵染色陽性,蒼白球輕度廣泛的星狀神經膠質增生及稀疏分布鐵陽性顆粒,中腦黑質色素神經元和腦橋藍斑色素神經元嚴重喪失,伴輕度星狀膠質增生,也有的表現為廣泛的橄欖腦橋小腦變性。而且,在皮質-紋狀體-蒼白球皮質環路均觀察到少突膠質細胞胞質內包涵體,此與基底核功能障礙有關。
臨床
 紋狀體黑質變性
紋狀體黑質變性一般於老年前期(25~68歲平均52歲)發病,散發性,隱襲發病,緩慢逐漸進展以帕金森綜合徵為首發症狀,在此背景上同時累及中樞神經系統其他部位,患者逐漸出現運動減少,運動不能(akinesia)、強直肢體和軀幹屈曲表情呆板,姿勢異常和步態變化構音障礙、吞咽困難和翻身困難等,約2/3患者在病程中可觀察到震顫,但震顫並不顯著,且75%~100%患者錐體外系症狀表現為非對稱的。一般發病較早並且對左鏇多巴治療無反應或反應極小,推測是由於攜帶多巴胺受體的紋狀體神經元減少造成的。Gonzalez等的研究認為與SND患者殼核多巴胺D2受體明顯減少有關,也有對左鏇多巴療效顯著者,此類患者更易誤診為帕金森病,但病後症狀進展較帕金森病快。
在錐體外系受損的基礎上可出現小腦性共濟失調症狀,或者先於帕金森綜合徵症狀出現,但多較晚且輕表現為平衡不穩,共濟失調等。
也可聯合出現OPCA症狀。另外有一半的患者有Shy-Drager綜合徵表現。許多患者有尿便控制障礙可能是由於骶部Onuf’s核神經元變性所致,也可有錐體束征的表現,由於黑質紋狀體功能障礙,部分患者呈現皮質下痴呆及神經心理障礙,但痴呆不常見,有的病例有巴賓斯基征,輕度肌萎縮,上視麻痹。病程呈進行性,一般為3~8年,平均死亡年齡57歲。
併發症:
隨病情發展出現多樣的症狀體徵可以是本病表現,也可以看作本病併發症。
另外,應注意繼發的肺部感染尿路感染等。
體症
 紋狀體黑質變性
紋狀體黑質變性(1)主要表現進行性肌強直、運動遲緩和步態障礙,病情發展到後期可導致自主神經損害、錐體束損害及(或)小腦損害。開始多為一側肢體僵硬、少動,病情逐漸發展至對側,導致動作緩慢、步態前沖、轉變姿勢困難、上肢固定、少擺動、講話慢及語音低沉等,但震顫很輕或缺如,可有位置性震顫,表現酷似Parkinson病,但大部分患者用左鏇多巴治療無效。
(2)隨著病情發展常出現步態不穩、共濟失調等小腦體徵,以及尿頻、尿急、尿失禁、尿瀦留、發汗障礙、體位性暈厥和性功能不全等自主神經功能障礙。少數可有錐體束征、雙眼向上凝視困難、肌陣攣、呼吸和睡眠障礙等。
(3)CT檢查可見雙側殼核低密度灶。MCI顯示殼核、蒼白球T2低信號,提示鐵沉著,早期病例可與Parkinson病區別。PET可顯示殼核和尾狀核18F-6-fluorodopar和11C-nomifensin攝取較正常減低,而Parkinson病這兩種顯像相對正常。
診斷
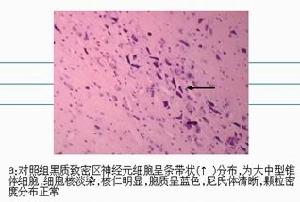 黑質神經元
黑質神經元診斷標準如下:
1,散發性成年潛隱起病的帕金森綜合徵,對左鏇多巴治療效果不佳。
2,具有明顯自主神經功能不全綜合徵。
3,出現小腦征。
4,出現錐體束征。
以上4項中,1必備,2,3兩項具備一項即可,4作為參考。
鑑別診斷:
臨床上有時與OPCA不易鑑別。Miwa報導SND患者55%有小腦症狀,而OPCA患者也有55%有帕金森綜合徵的表現,但SND患者發病年齡(60.7歲±8.7歲)較OPCA患者(55.4歲±7.9歲)晚。OPCA患者以小腦症狀為主,而SND以帕金森綜合徵症狀為主,首發症狀多為帕金森步態或震顫,無水平性眼運動障礙,另外顱腦MRI表現側重不同。
檢查
 紋狀體黑質變性
紋狀體黑質變性實驗室檢查:
1.直立實驗分別測量平臥位、坐位和直立位血壓站立2~3min內收縮壓下降多於30mmHg,舒張壓下降多於20mmHg心率無變化者為陽性。
2,血液生化檢查:血漿去甲腎上腺素含量測定、24h尿兒茶酚胺含量測定均明顯降低。
3,腦脊液檢查:除個別報導腦脊液內乙醯膽鹼酯酶降低外,大部分患者腦脊液均正常。
其它輔助檢查:
1,MR是最有價值的診斷方法,T2加權像顯示豆狀核低密度,紅核與黑質間正常存在的高信號降低,並顯示鐵沉積增加由於膠質細胞、膠質纖維增生及毛細血管增多使病變組織含水量增多,延長Tl、T2值,在Tl加權像呈低信號,T2加權像呈高信號。
O’Brien報告1例SND患者MRI顯示殼核信號減少,尤其是在後外側T2加權圖像低於蒼白球水平此外殼核明顯縮小,中腦由於黑質網狀部和紅核信號減小而顯示不清,作者認為SND的症狀似乎由於特殊結構功能的聯繫,殼萎縮與肌強直和對藥物不敏感有關肌強直的嚴重程度和殼信號減少程度密切相關。戴啟麟報導6例SND患者MRI檢查均異常。2例雙側外囊區對稱出現白質退行性變4例出現雙側殼核不規則T1W1低信號,T2W1高信號其中1例同時伴腦幹小腦萎縮,而6例CT檢查均正常。
2,PET研究表明紋狀體額葉小腦和腦幹葡萄糖代謝障礙,是由於功能性神經元成分缺失造成的。
治療
治療無特效療法,主要用支持及對症治療網。有人認為左鏇多巴替代療法至少對35%的患者有暫時性效果。目前,歐洲的研究者正致力於開一種神經保護療法,正在進行的研究,是試驗通過紋狀體移植的神經移植療法是否是一個可誘導左鏇多巴(L-多巴)的臨床相關反應的有效療法(WenningGK健康搜尋2001),神經移植對此病引起的症狀、早期殘廢和平均壽命的明顯降低可能有影響。
預防
 紋狀體黑質變性
紋狀體黑質變性預後:
病程呈進行性一般為3~8年。
平均死亡年齡57歲。
預防:
尚無有效的預防方法。
對症處理是臨床醫療護理的重要內容。
區別
 紋狀體黑質變性
紋狀體黑質變性研究多系統萎縮的紋狀體黑質變性型(MSA-P)與原發性帕金森病(IPD)在18F-FDGPET顯像上的差異。方法:11例臨床診斷多系統萎縮紋狀體黑質變性型和25例病程匹配的臨床診斷原發性帕金森病患者進行頭部18F-FDGPET顯像檢查(標記物量為2.2-2.6毫居),運用放射自顯影公式計算局部腦葡萄糖代謝率,以評價組織活性。用SPM法進行圖象分析和處理。比較兩組患者的基底節區的局部腦葡萄糖代謝率(rCMRGlc),以及殼核局部腦葡萄糖代謝率前後部分、左右的對稱性。用全腦葡萄糖代謝率(gCMRGlc)除以rCMRGlc以對rCMRGlc進行標準化。結果:與IPD組相比,MSA-P組殼核總rCMRGlc、殼核前部rCMRGlc、殼核後部rCMRGlc明顯降低(P0.05)。IPD組在症狀較重對側的殼核後部rCMRGlc低於對側(P0.05)。這些結果不依賴於整個腦葡萄糖代謝率水平,因為rCMRGlc值經標準化後仍可得到相同的結果。
病例
臨床資料:男1例,女3例;年齡50~64歲,平均55歲。均無家族遺傳史,均為隱襲性發病,緩慢逐漸進展,病後均表現為走路不穩、動作緩慢,病程中均一度出現肢體輕微震顫。表現走路易跌倒3例,尿失禁、便秘3例,尿瀦留1例,下半身不出汗3例,流涎1例,智力減退4例。入院前服美多巴等抗PD藥物效果均不明顯。入院檢查,呈面具臉3例,齒輪樣肌張力、主動運動減少及動作緩慢4例。指鼻、跟膝脛試驗及昂白征均為陽性,雙巴賓斯基征陽性2例,腱反射活躍2例,感覺檢查均正常。從發病至死亡時間1.5~6年,平均3.6年。腦電圖檢查均正常,檢查誘發電位2例示腦幹功能傳導障礙,頭CT示輕度腦萎縮3例,其中腦幹、小腦、基底節萎縮2例,入院後給美多巴等抗PD治療,病情漸加重,均死於肺炎及感染性休克。
