
原理
反向PCR可用於研究與已知DNA區段相連線的未知染色體序列,因此又可稱為染色體緩移或染色體步移。這時選擇的引物雖然與核心DNA區兩末端序列互補,但兩引物3’端是相互反向的。擴增前先用限制性內切酶酶切樣品DNA,然後用DNA連線酶連線成一個環狀DNA分子,通過反向PCR擴增引物的上游片段和下游片段;現已製備了酵母人工染色體(YAC)大的線狀DNA片段的雜交探針,這對於轉座子插入序列的確定和基因庫染色體上DNA片段序列的識別十分重要。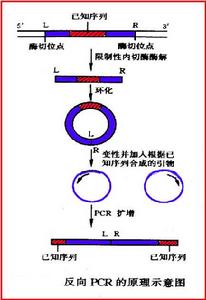
不足之處
該方法的不足是:①需要從許多酶中選擇限制酶,或者說必須選擇一種合適的酶進行酶切才能得到合理大小的DNA片段。這種選擇不能在非酶切位點切斷靶DNA。②大多數有核基因組含有大量中度和高度重複序列,而在YAC或Cosmid中的未知功能序列中有時也會有這些序列,這樣,通過反向PCR得到的探針就有可能與多個基因序列雜交。其他用途
利用反向PCR可對未知序列擴增後進行分析,探索鄰接已知DNA片段的序列,並可將僅知部分序列的全長cDNA進行分子克隆,建立全長的DNA探針。適用於基因遊走、轉位因子和已知序列DNA旁側病毒整合位點分析等研究。用傳統的緩衝液和其他提供者推薦的條件裂解DNA。反向PCR所擴增的片段的大小由 PCR擴增片段的大小決定,目前,PCR擴增的實際上限為3-4kb。在許多情況下,首先 需要進行Southern雜交來確定內切酶用以產生大小適於環化及反向PCR的片段的末端 片段。能裂解核心區的內切酶使反向PCR只能擴增引物所定模板(依賴於引物)的上游 或上游區,而不裂解核心區的酶則使兩上邊側序列都擴增,並帶有由內切酶和環化類 型決定的接點(例如,互補突頭連線與鈍頭連線)。對於擴增左翼或右翼序列,初試時 最好靠近識別上個鹼基位位的酶,並已知在核心區有其方便的裂解位點。如果用反向 PCR從含有大量不同的克隆片段的同一載體中探測雜交探針,建議事先在載體中引入 合適的酶切位點。
用T4連線酶在稀DNA濃度下環化更容易形成單環。在一些實驗中,為產生對反向 PCR大小適當的DNA片段需要兩種內切酶,但這樣所產生的片段末端則不適於連線,環 化前需用Klenow或噬菌體T4DNA聚合酶修理(鈍化)。連線前,需用酚或熱變性使內切 酶失活。
聚合酶鏈反應條件與經典所用的相同,例如,94℃-30秒變性,58℃-30秒引物退火, Taq聚合酶70℃延伸3分鐘,進行30個循環。可改變PCR條件以生產特異產物。將反向 PCR用於測序時,與核心區末端後部結合的擴增引物更為有用,它使測序引物擴增部 分的核心序列與未知邊側序列間的接點更近,減少了擴增引物的干擾。
