症狀體徵
 克羅伊茨費爾特-雅各布病
克羅伊茨費爾特-雅各布病有將其分為兩種類型:經典型和新變異型。分別闡述如下:
 朊蛋白
朊蛋白2.新變異型臨床表現多在青年期發病,起病亞急性或緩慢發病,早期出現精神症狀:表情淡漠、情緒低沉、抑鬱焦慮或焦慮不安,全身乏力萎靡。發病數周或數月出現進行性共濟失調,認知障礙出現較晚,可有肌陣攣抽動、舞蹈樣動作,晚期出現記憶力喪失,進行性痴呆。
神經檢查體徵:步態不穩、步態蹣跚、昂伯征,小腦性共濟失調,少數病人可見面部或軀體感覺障礙,可見錐體束征和錐體外束征,痴呆、緘默不語,最終多在2年以後衰竭死亡。
病因病理
疾病病因
 克羅伊茨費爾特-雅各布病
克羅伊茨費爾特-雅各布病病理生理
因瘙癢病特異蛋白為非常規因子,不具有免疫原性,進入機體後不能刺激機體進行細胞的體液免疫反應,因而一旦感染便不能清除病原,使大腦皮質出現病理性海綿狀改變,神經細胞減少、變性和星形細胞增生改變等。經典型早期一般僅見輕度腦萎縮。鏡下顯示大腦皮質灰質可見大小不等、圓形和卵圓形空泡呈海綿狀改變,神經細胞變性減少和脫失伴隨星形細胞增生,無炎性細胞浸潤。PrP免疫組化染色陽性,剛果紅染色偶見澱粉樣斑。電鏡所見膜狀空泡變性和羊瘙癢症相關纖維(SAF)。
晚期則見明顯腦池變寬、腦回扁平嚴重腦萎縮,無炎性滲出和局灶性腦病變。病理性海綿狀改變主要見於大腦皮質,其分布以顳、額、枕葉為最著,頂葉和中央回則最輕,基底節殼核、尾狀核和丘腦病變明顯,而蒼白球大細胞較輕,小腦、中腦、腦橋和延髓亦可見海綿狀改變、神經細胞減少變性和星形細胞增生。脊髓前角亦可受累但較輕。病程短者海綿狀改變重,病程長者神經細胞變性和星形細胞增生明顯。
診斷檢查
診斷
 克羅伊茨費爾特-雅各布病
克羅伊茨費爾特-雅各布病腦脊液化驗多正常,總蛋白偶見增多,但免疫球蛋白不增多。氣腦造影正常或腦室擴大,可存在大腦和小腦皮質萎縮的徵象。腦電圖往往有明顯異常,開始可出現瀰漫或局灶性慢波,以後可見尖波或慢峰形波,最後在皮質抑制活動背景上出現特徵性三相同步複合尖波,對臨床診斷有重要價值。頭顱CT可見皮質萎縮和腦室擴大。病理改變的特點是海綿狀改變、神經細胞變性或脫失和星形細胞增生等,有助於確診。
1.經典型散發性克-雅氏病診斷標準
(1)肯定(definite)CJD診斷:
①具有典型臨床症狀和體徵(錐體束征和錐體外束征)。
②具有連續觀察腦電圖出現三相波和周期性同步發放(PSD)。
③腦組織活檢得到病理支持:
A.典型的海綿狀改變。
B.澱粉樣蛋白斑沉積,PrP免疫組化(+)。
C.電鏡觀察:膜狀空泡變性和羊瘙癢症相關纖維(SAF)。
(2)可能(Probable)CJD診斷:
①進行性痴呆。
②典型腦電圖三相波和PSD。
③腦脊液14-3-s蛋白陽性,臨床病程<2年。
④具有以下臨床症狀4項中2項者:
A.肌陣攣抽動。
B.視覺或小腦障礙。
C.錐體束征/錐體外束征。
D.無動性緘默。
(3)可疑(Possible)CJD診斷:與可能(Probable)CJD臨床表現相似;但無典型腦電圖所見或有不典型腦電圖改變;病程短於2年者。
2.新變異型CJD診斷標準
(1)臨床表現:
①精神異常、抑鬱焦慮症狀。
②發病數周或數月出現小腦共濟失調。
③記憶力喪失,晚期出現痴呆。
④肌陣攣抽動、舞蹈樣症狀和錐體外束征。
⑤肢體乏力、面部和肢體感覺異常,錐體束征。
⑥腦電圖無特徵性改變。
(2)確診必須病理證實:
①可見豐富Kuru型澱粉樣斑圍以空泡。
②海綿樣改變以基底節區為明顯。
③丘腦可見膠質增生明顯。
④PrP沉積見於大腦和小腦分子層。
 克羅伊茨費爾特-雅各布病
克羅伊茨費爾特-雅各布病1.實驗室檢查
腦脊液常規、生化檢查除少數病例有蛋白輕度升高(一般50~60mg/dl)外,無異常改變。免疫球蛋白(Ig)和寡克隆區帶均無異常表達。檢測14-3-s蛋白陽性具有輔助診斷價值。
2.其他輔助檢查
1.腦電圖往往有明顯異常,腦電圖檢查對經典型克-雅氏病具有重要輔助價值。其特殊性改變:早期僅見散在α波減少,相繼出現α波減少消失,出現θ波和δ波。病情進展則α波消失,出現瀰漫或局灶性慢波,以後可見尖波或慢峰形波,棘-慢波綜合波,最後在皮質抑制活動背景上出現特徵性三相同步複合尖波,最終腦電圖背景電靜息和周期性同步發放(PSD)。而新變異型克-雅氏病則無此特殊征性腦電圖改變。開始可出現對臨床診斷有重要價值。
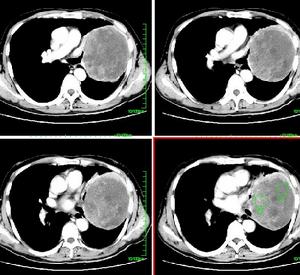
2.影像學檢查頭顱CT、MRI早期無異常改變,病情進展期可見普遍性腦萎縮,腦池寬、腦回小和腦室擴大,多數(80%)無局灶性改變。發現少數病例MRI可見雙側底節、丘腦後部長T2信號。有時較不明顯,如用質子像、Flair和彌散MRI可見明顯增強信號。具有診斷意義。
3.氣腦造影正常或腦室擴大,可存在大腦和小腦皮質萎縮的徵象。
鑑別診斷
 克羅伊茨費爾特-雅各布病
克羅伊茨費爾特-雅各布病應與老年期中樞神經系統變性病痴呆鑑別:
1.阿爾采默病呈隱襲起病緩慢進展性痴呆,CT、MRI可見額顳葉萎縮。多在老年前期和老年期發病,病程長達5~10年以上,病理改變為老年斑和神經纖維纏結。
2.匹克病在老年前期起病,進展緩慢,多有家族史,以腦葉萎縮為特點,人格改變,呈進行性痴呆,CT、MRI可見額、顳葉明顯萎縮。
3.亨廷頓舞蹈病(HuntingtonChorea)中年起病,有明顯家族史,臨床表現舞蹈征伴進行性痴呆,CT、MRI可見尾狀核萎縮、側腦室擴大,病程可長達10~30年以上。
4.帕金森病痴呆帕金森病並發痴呆者少見,只占該病3%~5%。起病緩慢,以動作緩慢、肌強直、靜止性震顫三聯征在先,痴呆出現在後,且痴呆常發生在該病的晚期,病程長達4~10年以上。
治療預防
 克羅伊茨費爾特-雅各布病
克羅伊茨費爾特-雅各布病尚無特異性有效的治療方法。但仍需給予良好的護理和有效的對症治療及支持療法。對抽搐、肌陣攣和興奮躁動仍應給予積極治療為宜。據文獻報導,金剛烷胺200mg/d,3次/d,口服,對本病有效,但也有無效者。
併發症
和其他重症疾病一樣,易罹患各系統繼發感染,尤以墜積性肺炎和泌尿系感染多發。
預後
本病85%為散發,預後不良,多數發病後3~12個月死亡。絕大多數在2年內死亡。
預防
實驗證明病人腦組織接種傳遞發病率高,潛伏期長,但其傳染途徑未明。應積極採取預防措施。
1.對醫源性傳染如角膜移植、骨髓移植、生物製劑、生長激素等嚴加監控,杜絕傳染。
2.雖然對於病人一般接觸未見到傳染致病的報導,但對有創傷檢查如取血、腰穿取腦脊液、頭皮穿刺、腦組織活檢等,要嚴格執行消毒隔離杜絕傳染。
(1)儘量使用一次性器械如注射器、針頭,用後處理嚴加管理,以焚燒為宜。
(2)對不能一次性使用的器械,可設專用於CJD腦活檢器械,不用於一般病人手術,採取特殊消毒措施,132℃高壓消毒60min以上或5%漂白粉液或2mol/l氫氧化鈉浸泡60min。
(3)病人的活檢或屍檢腦組織嚴加管理,對其污染物的處理一定要焚燒。
流行病學
克羅伊茨費爾特-雅各布病性痴呆發病率每年1/100萬,以色列、利比亞人發病率最高,為一般人群的10~20倍。男女發病率相等或男性稍高於女性,發病年齡50~70(65)歲。但可在成年的各階段發病,多為散發病例,經典型克-雅氏病85%以上為散發性,病因和傳染途徑不十分清楚,10%~15%為家族性,還有少數為醫源性傳遞如角膜移植、硬膜移植,顱腦手術、皮質電極檢查,也有接受生物製劑治療傳遞如人生長激素治療而發病者。
