流行病學
 顯微鏡下多血管炎
顯微鏡下多血管炎顯微鏡下多血管炎在任何年齡都可發病,但以40~50歲最常見,發病率為1/10萬~3/10萬人,男性發病率略高於女性,男女比為1~1.8∶1。
病因顯微鏡下多血管炎的病因仍不清楚,有資料表明與病人體內的免疫異常有關。
發病機制
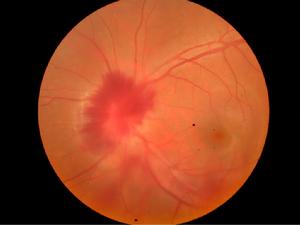 顯微鏡下的症狀
顯微鏡下的症狀2.病理 顯微鏡下多血管炎在組織病理學上表現為以微小靜脈、微小動脈和毛細血管受累為主但也可有中等大小動脈受累的血管炎。主要表現為局灶性壞死性的全層血管炎,正常的血管結構往往被破壞病變部位可見纖維素樣壞死和多種細胞[如多形核白細胞、淋巴細胞(主要為CD4+細胞)及嗜酸性粒細胞]的浸潤。同一病人的血管炎症可有不同的階段,活動性和已癒合的血管損傷可同時存在癒合部位主要表現為纖維組織和內皮細胞的增生,可造成血管的阻塞。
顯微鏡下多血管炎在病理學上與結節性多動脈炎的表現無明顯差別,但顯微鏡下多血管炎在腎臟的病變除見有腎臟小血管的炎症改變以外,主要表現為壞死性新月體形成型腎小球腎炎是它的特徵性改變之一。另一較有特徵性的改變是肺毛細血管炎。
在20世紀初期,顯微鏡下多血管炎的發生一直被認為可能與過敏因素如異源性蛋白質、細菌和藥物等有關。顯微鏡下多血管炎的發病機制目前並不十分清楚。可能與下列機制有一定關係:①抗中性粒細胞胞質抗體和中性白細胞介導的內皮細胞損傷;②抗內皮細胞抗體的作用;③細胞免疫介導的內皮細胞損傷。
(1)抗中性粒細胞胞質抗體:許多顯微鏡下多血管炎血清中存在抗中性粒細胞胞質抗體,顯微鏡下多血管炎與韋格納肉芽腫一樣,是一種無免疫複合物沉積的血管炎,因此抗原-抗體免疫複合物介導的血管損傷在顯微鏡下多血管炎的發病機制中意義並不大。目前認為抗中性粒細胞胞質抗體可能通過介導中性粒細胞的活化而發揮作用:感染等各種因素使顯微鏡下多血管炎患者血清中各種炎性細胞因子如腫瘤壞死因子-α和白細胞介素-1的水平升高,它們誘導黏附分子的表達這使得多形核白細胞易黏附於血管內皮。同時還能誘導多形核白細胞內的蛋白酶-3從胞質內的嗜苯胺藍顆粒轉移到細胞表面。當抗中性粒細胞胞質抗體與白細胞表面的蛋白酶-3結合時可激活白細胞引起多形核白細胞脫顆粒釋放活性氧物質及溶酶體酶等,導致周圍血管損傷和壞死。另一方面腫瘤壞死因子-α等細胞因子還能激活血管內皮細胞,活化的內皮細胞表達髓過氧化物酶、蛋白酶-3等抗原,使抗中性粒細胞胞質抗體通過與其特異性抗原直接結合到內皮細胞上,經抗體介導的細胞毒殺傷途徑溶解血管內皮引起血管的損傷。
(2)抗內皮細胞抗體:抗內皮細胞抗體存在於許多系統性血管炎包括顯微鏡下多血管炎中,但抗內皮細胞抗體在顯微鏡下多血管炎中的作用並不十分清楚。有研究發現許多抗內皮細胞抗體並無致病性。抗內皮細胞抗體導致內皮細胞的損傷,其機制可能是通過補體介導的溶解途徑和抗體介導的細胞毒作用而實現的。
臨床表現
 治療用藥
治療用藥1.全身症狀 顯微鏡下多血管炎患者在就診時常伴有一般全身情況,包括發熱、乏力、厭食、關節痛和體重減輕。
2.皮膚表現 MPA可出現各種皮疹以紫癜和高出皮面的充血性斑丘疹多見。皮疹可單獨出現,也可和其他臨床症狀同時出現,其病理多為白細胞破碎性血管炎。除皮疹外,MPA患者還可出現網狀青斑、皮膚潰瘍、皮膚壞死、壞疽以及肢端缺血、壞死性結節、蕁麻疹和血管炎相關的蕁麻疹常持續24h以上。
3.腎臟損害 腎臟損害是MPA最常見的臨床表現,病變表現差異很大,極少數患者可無腎臟病變。多數患者出現蛋白尿、血尿各種管型水腫和腎性高血壓等;部分患者出現腎功能不全可進行性惡化致腎功能衰竭。25%~45%的患者最終需血液透析治療。
MPA的腎臟病理為壞死性腎小球腎炎,其特徵為節段性壞死伴新月體形成,很少或無毛細血管內皮細胞增殖。腎小球組織學很少或無免疫複合物沉積電鏡下很少或無電子緻密物沉積。以上特點和其他的免疫複合物介導的腎小球腎炎以及抗腎小球基底膜抗體介導的Goodpasture綜合徵不同,但和韋格納肉芽腫的腎臟病變以及特發性的急進性腎小球腎炎有時不易鑑別。MPA光鏡下的病理改變見。
4.肺部損害 約一半的MPA患者有肺部損害發生肺泡毛細血管炎,12%~29%的患者有瀰漫性肺泡出血。查體可見呼吸窘迫征肺部可聞及囉音。由於瀰漫性的肺間質改變和炎症細胞的肺部浸潤,約1/3的患者出現咳嗽、咯血、貧血,其中大量的肺出血可導致呼吸困難,甚至死亡。部分患者可在瀰漫性肺泡出血的基礎上出現肺間質纖維化。
5.神經系統 20%~30%MPA患者有神經系統損害的症狀,其中約57%出現多發性單神經炎或多神經病變,另約11%的患者可有中樞神經系統受累,常表現為癲癇發作。
6.消化系統 消化道也可被累及,表現為消化道出血、胰腺炎以及由腸道缺血引起的腹痛。嚴重時可由於胃腸道的小血管炎和血栓形成造成缺血,導致腸穿孔。
7.心血管系統 MPA亦可累及心血管系統,患者可出現胸痛和心衰症狀,臨床可見高血壓、心肌梗死以及心包炎。
8.其他 部分患者也有耳鼻喉的表現,如鼻竇炎,此時較易與韋格納肉芽腫相混淆。少數患者還可有關節炎關節痛和睪丸炎所致的睪丸痛。
眼部症狀包括眼部紅腫和疼痛以及視力下降,眼科檢查表現為視網膜出血鞏膜炎以及葡萄膜炎。
併發症MPA病理特徵和臨床表現為多系統損害,其併發症亦呈多樣性。
診斷本病診斷尚無統一標準,如出現系統性損害並有肺部受累腎臟受累及出現高出皮面的紫癜應考慮MPA的診斷尤其是同時具有p-ANCA陽性者。腎活檢及皮膚或其他內臟活檢有利於MPA的診斷。部分患者需除外感染性心內膜炎。
 觀察
觀察確定診斷之前,需與結節性多動脈炎和韋格納肉芽腫相鑑別。
1.結節性多動脈炎(PAN) 以往MPA屬於PAN的一種類型,隨著疾病認識的不斷深入,發現二者臨床表現並不完全相同,故1993年的關於血管炎的教會山會議(Chapel Hill consensus conference)把MPA單獨列為一種疾病。根據新的定義,PAN是累及中動脈以及小動脈的壞死性炎症不伴有腎小球腎炎或微小動脈毛細血管或微小靜脈炎症;而MPA是主要累及小血管的壞死性血管炎,很少或無免疫複合物沉積,其中壞死性腎小球腎炎很多見,肺毛細血管炎也常發生。
2.韋格納肉芽腫(WG) WG為小動脈和小靜脈的血管炎,以上下呼吸道和腎臟病變三聯征為主要臨床特點,c-ANCA陽性多見活檢病理示小血管壁或其周圍有嗜中性粒細胞浸潤,並有壞死性肉芽腫形成。而MPA很少累及上呼吸道,主要為p-ANCA陽性,一般無肉芽腫形成。
3.肺出血-腎炎綜合徵(Goodpasture syndrome) Goodpasture綜合徵也稱為抗腎小球基底膜抗體腎炎伴肺出血(anti-GMB disease withpulmonary hemorrhage),是由於肺泡和腎小球基底膜受損而致病,包括反覆瀰漫性肺出血腎小球腎炎以及循環抗腎小球基底膜抗體(anti-GBM)三聯征,臨床表現為反覆瀰漫性肺出血、貧血以及腎出血(血尿)。肺及腎活檢經免疫螢光鏡檢查可見抗基底膜抗體的IgG及C3沿肺泡壁以及腎小球的毛細血管壁呈連續均勻線狀沉積。血循環中檢出抗基底膜抗體是診斷本病的重要依據。
檢查
 中醫治療
中醫治療1.常規檢查 在MPA中,反映急性期炎症的指標如ESR、CRP升高,部分患者有貧血白細胞和血小板增多。累及腎臟時出現蛋白尿、鏡下血尿和紅細胞管型,血清肌酐和尿素氮水平升高。
2.免疫學檢查 C3和C4水平正常。約80%的MPA患者抗中性粒細胞胞質抗體(ANCA)陽性,是MPA的重要診斷依據,其中約60%MPO-ANCA(p-ANCA)陽性,肺受累及者常有此抗體,另有約40%的患者為PR3-ANCA(c- ANCA)陽性。約40%的患者可查到抗心磷脂抗體(ACL)少部分患者ANA、RF陽性。北京協和醫院1995~2001年確診的16例MPA患者中ANCA陽性的有13例(81.2%),其中11例(84.6%)為P-ANCA陽性,3例(23.1%)c-ANCA陽性的患者中有1例(7.7%)同時p-ANCA陽性。
胸片:早期可發現無特徵性的雙側不規則的結節片狀陰影或小泡狀浸潤影,肺空洞少見,可見繼發於肺泡毛細血管炎和肺出血的瀰漫性肺實質浸潤影,中晚期可出現肺間質纖維化。
治療
 顯微鏡下多血管炎
顯微鏡下多血管炎MPA的治療可以分為3個階段,第1階段為誘導緩解;第2階段為維持緩解,此階段可以中等量潑尼松治療並維持環磷醯胺(CTX)治療12個月,或換用硫唑嘌呤、甲氨蝶呤等DMARDs維持緩解;第3階段為治療復發,可採用與誘導緩解同樣的治療方案金黃色葡萄球菌的定植可能和MPA的復發有一定的關係,因此服用磺胺類抗生素對防止復發有一定效果。
對於伴有肺出血的肺泡毛細血管炎危及生命的患者應聯合治療或行血漿置換治療。糖皮質激素加環磷醯胺應作為首選方案。
1.糖皮質激素 糖皮質激素是治療MPA、誘導緩解的一線用藥。為儘快誘導緩解,可採用甲潑尼龍(甲基潑尼松龍)衝擊治療,劑量為7mg/(kg·d),連用3天,然後改用潑尼松逐漸減量。潑尼松初始劑量為40~60mg/d待ESR降至正常,患者症狀消失後開始減量,每1~2周減量5~10mg。劑量減至15mg時,減量宜慢。
初治者尤其是有肺、腎損害的,常用潑尼松60mg/d,並聯合用環磷醯胺,療程要長,停藥後,仍有約25%的患者平均在24個月內復發。
2.免疫抑制劑
(1)環磷醯胺(CTX):環磷醯胺應作為首選治療,劑量為靜脈給藥0.5~1g/(m2·月),或0.2g靜脈推注隔日一次,或0.1g口服1次/d。用藥過程中根據白細胞計數調整劑量,用藥時間要長,通常達12個月。
(2)甲氨蝶呤(MTX):MTX可以抑制炎症,減輕炎症症狀。劑量為10~25mg/周,口服、肌注和靜脈注射均可。
(3)硫唑嘌呤:現常用的劑型為硫唑嘌呤(依木蘭),是嘌呤代謝的拮抗劑,可以抑制DNA和RNA的合成,從而降低免疫細胞的增生,下調免疫活性劑量為1mg/(kg·d),常用每天50~100mg。用藥6~8周后,如初始劑量效果不佳,在無嚴重不良反應的情況下可以加大劑量以0.5mg/(kg·d)的速度增加,必要時每4周可以調整劑量總劑量勿超過2.5mg/(kg·d)。
3.靜脈用人血丙種球蛋白(丙種球蛋白) 對環磷醯胺治療反應不佳的患者可選用靜脈用人血丙種球蛋白,可明顯改善肺、腎損害的臨床症狀,抗獨特型抗體可能是有效的作用機制。IVIg的劑量為400mg/(kg·d)連用5~7天國內常用劑量為20g/d。。
預後預防
 示意圖
示意圖90%的MPA患者經治療能得到改善,75%的患者能完全緩解約30%的患者在1~2年後復發。本病治療後的2年和5年生存率大約為75%和74%與PAN相似本病的主要死亡原因是不能控制的病情活動腎功能衰竭和繼發感染以及肺臟受累。北京協和醫院確診的MPA中有2例在住院期間死亡,病因為急進性腎小球性腎炎(rapidly progressive glomerulonephritisRPGN)、彌散性肺泡出血和敗血症。疾病過程中應密切監測ESR水平,MPA中ANCA的滴度與病情活動相關性較差。
預防
1.一級預防。
(1)防止可能的誘因,居室不宜過冷和潮濕,溫度要適宜。
(2)預防感染加強鍛鍊身體,增強體質,提高自身免疫功能,生活規律。
(3)加強營養,不可貪冷飲和過食肥甘厚味之品,忌食辛辣食物和忌菸酒。
2.二級預防。
(1)早期診斷較難,凡是年輕人尤其是女性,有下列一種情況者,應考慮本病,全身發熱,關節或肌痛,單側或雙側肢體出現缺血症狀,頭部缺血症狀頑固性高血壓症狀,血管雜音等症,應及早就醫明確診斷。
(2)綜合治療,減少併發症,改善預後。
①內科治療:包括控制感染,糖皮質激素,改善微循環,抗凝降壓藥及中醫辨證治療。
②外科治療:有經皮腔內血管成形術動脈轉流術、瓣膜修復術及腎臟切除術等。
3.三級預防 目前大動脈炎的治療尚無特效藥物。中醫藥具有調節免疫清熱解毒、活血化瘀的功效。此外仍需加強全身營養、身體鍛鍊、生活規律、勞逸結合、心情舒暢等。
