血紅蛋白病(hemoglobinopathy)是由於血紅蛋白分子結構異常(異常血紅蛋白病),或珠蛋白肽鏈合成速率異常(珠蛋白生成障礙性貧血,又稱海洋性貧血)所引起的一組遺傳性血液病。臨床可表現溶血性貧血、高鐵血紅蛋白血症或因血紅蛋白氧親和力增高或減低而引起組織缺氧或代償性紅細胞增多所致紫紺。
內容
血紅蛋白病
【概述】
血紅蛋白是一種結合蛋白,分子量64,000,由珠蛋白和血紅素構成。血紅素由原卟啉與亞鐵原子組成,每一個珠蛋白分子有二對肽鏈,一對是α鏈,由141個胺基酸殘基構成,含較多組氨酸,其中α87位(即F8)組氨酸與血紅素鐵的結合,在運氧中具重要生理作用。另一對是非α鏈,有β、γ、δ、ξ(結構與α鏈相似)及ε5種;後2種與α鏈、γ-鏈分別組成胚胎早期(妊娠3月以內)血紅蛋白、HbGower-1(ζ2ε2)、HbGower-2(α2ε2)、HbPortland(ζ2γ2)。β鏈含146個胺基酸殘基、β93半胱氨酸易被氧化產生混合二硫化物及其它硫醚類物質,可降低血紅蛋白穩定性。δ鏈亦由146個胺基酸殘基組成,僅10個胺基酸與β鏈不同。由於δ鏈中第22位丙氨酸置換了β22谷氨酸,第116位精氨酸置換了β116組氨酸,因此δ鏈的正電荷大於β鏈,HbA2(α2δ2)等電點升高,電泳時靠近負極。γ鏈雖由146個胺基酸組成,但與β鏈有39個胺基酸不同,且含有4個異亮氨酸,為α、β與δ鏈所缺如,因此可用分析異亮氨酸方法以測定HbF(α2γ2)含量。正常人有二種γ鏈、Gr-R136為甘氨酸,Ar-r136為丙氨酸,說明控制γ鏈生物合成的基因位點不止一個。初生時Gr與Ar的比例是3∶1,兒童和成人二者之比為2∶3。每一條肽鏈和一個血紅素連線,構成一個血紅蛋白單體。人類血紅蛋白是由二對(4條)血紅蛋白單體聚合而成的四聚體。不同類型的血紅蛋白珠蛋白結構略有不同,但血紅素均相同。
 血紅蛋白病
血紅蛋白病血紅蛋白的四級結構:由胺基酸順序排列的肽鏈結構稱為血紅蛋白的一級結構。肽鏈中的胺基酸可分為親水的極化胺基酸(其側鏈為羧基、氨基),與非極化的胺基酸(其側鏈是芳香族)。肽鏈中的各種胺基酸的側鏈相互拉緊形成α螺鏇,螺鏇形節段間由短而非螺鏇形節段相連。螺鏇形節段從N端-C端分別以A-H表示,非螺鏇形節段用AB、CD等表示,稱為血紅蛋白的二級結構。血紅素的鐵原子有6個配位鍵,第5個配位鍵結合在肽鏈F段第8位胺基酸上(即α鏈第87位或β鏈第92位組氨酸的咪唑基上),第6個配位鍵結合氧,並間接結合在肽鏈E段的第7位胺基酸上(即α鏈第58位或β-鏈第63位組氨酸的咪唑基上),使肽鏈圍繞血紅素為中心,構成內外二層螺鏇狀蛇形盤曲的三維空間結構,稱為三級結構(圖20-12)。親水胺基酸分布於外層,使血紅蛋白能溶於水而不致沉澱;疏水胺基酸分布於內層,使水分子不能進入血紅素腔內部,避免血紅素的Fe2+氧化為Fe3+。四個血紅蛋白單體(肽鏈三級結構加血紅素),按一定的空間關係結合成四聚體,如HbA(或HbA1,α2β2)、HbA2(α2δ2)及HbF(α2γ2),稱異質型四聚體;由二對同樣的三級結構血紅蛋白單體結合成的四聚體,如HbH(β4)及HbBart(γ4),稱為同質型四聚體。以上所述四聚體為血紅蛋白四級結構。通過X線衍射研究四聚體的空間關係,發現α1β1及α2β2的接觸面較大,相互移動度較小,疏水,有利於血紅蛋白分子構型的穩定性。α1β2及α2β1接觸面小而不牢固,移動度大,有利於血紅蛋白對氧的正常攝取與釋放。四聚體解離,首先離解為α1β1及α2β2。綜上所述,血紅蛋白與分子的外表結構必需完整,帶有負電荷;α、β鏈結合部位要固定,包圍血紅素腔的胺基酸順序排列應完整,否則血紅蛋白就不能維持分子結構穩定性及正常運輸氧生理功能,並易遭破壞。
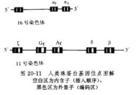
正常人出生後有三種血紅蛋白:①血紅蛋白A(HbA),由一對α鏈和一對β鏈組成(α2β2),為正常人主要血紅蛋白,占血紅蛋白總量的95%以上。胚胎二個月時HbA即有少量出現,初生時占10%~40%,出生6個月後即達成人水平;②血紅蛋白A2(HbA2),由一對α鏈和一對δ鏈組成(α2δ2)。自出生6~12個月起,占血紅蛋白的2~3%;③胎兒血紅蛋白(HbF)由一對α鏈和一對γ鏈組成(α2γ2),初生時占體內血紅蛋白的70%~90%,以後漸減。至生後6月,含量降至血紅蛋白總量的1%左右。血紅蛋白的不同肽鏈是由不同的遺傳基因控制的,α鏈基因位於第16號染色體,β、δ、γ鏈基因位於第11號染色體,呈連鎖關係(圖20-11)。α珠蛋白基因的缺失或缺陷,導致α珠蛋白鏈合成減少或缺乏,稱為α海洋性貧血。β珠蛋白基因缺陷,導致β珠蛋白鏈合成減少或缺乏,稱為β海洋性貧血。珠蛋白基因突變而致肽鏈的單個或多個胺基酸替代或缺如,導致珠蛋白分子結構改變,稱為異常血紅蛋白。全世界範圍內經結構分析證實的異常血紅蛋白日益增多,至90年代初期已達600多種,但僅不到1/3的異常血紅蛋白伴有臨床症狀。世界衛生組織估計,全球約有1.5億人攜帶血紅蛋白病基因,並已將血紅蛋白病列為嚴重危害人類健康的6種常見病之一。異常血紅蛋白病在我國以雲南、貴州、廣西、新疆等地發病率較高,現已發現異常血紅蛋白67種,包括α鏈(34種)、β鏈(26種)、γ鏈(4種)等異常,其中19種為我國首見。海洋性貧血多發於華南及西南地區。根據近10年來我國28個省市、自治區近100萬人口的普查資料,異常血紅蛋白病的發病率為0.33%,α海洋性貧血的發病率為2.64%,β海洋性貧血的發病率為0.66%。
【分子遺傳學】
血紅蛋白的分子遺傳變化,大致可歸納為以下6類:
(一)單個鹼基替代 由於遺傳密碼中單個鹼基替代,導致由該鹼基決定的胺基酸發生相應的變化,形成肽鏈中單個胺基酸置換的異常血紅蛋白,例如HbS、HbC等。目前發現的異常血紅蛋白中,以本類型最多見,約占90%。
(二)終止密碼的突變 因終止密碼(UAA、UAG)的變異,使珠蛋白肽鏈不在正常的位置終止,導致肽鏈延長或縮短,如Hb McKees Rock的β鏈第145位胺基酸的鹼基由UAU變為UAA(終止密碼),使β鏈提前結束,僅含144個胺基酸。又如Hb ConstantSpring α鏈第142位終止密碼UAA變為CAA,直至第173位才出現終止密碼,因此Hb Constant Spring α鏈比正常α鏈多32個胺基酸。
(三)移碼突變 如正常血紅蛋白肽鏈遺傳密碼中,嵌入或缺失1~2個鹼基,使正常三聯密碼子鹼基成分發生改變,如HbTak為β鏈第147位終止密碼UAA前插入AC,使UAA→ACU蘇氨酸,而致β鏈延長至第157位胺基酸,比正常β鏈多11個胺基酸。
(四)密碼子缺失或插入 生殖細胞減數分裂時,聯合中的染色體發生錯配或不等交換,形成兩種珠蛋白基因。一種失去一部分密碼子,合成缺失部分胺基酸的肽鏈,如HbLyon,β鏈第17-18位缺失賴、纈氨酸。另一條染色單體上卻嵌入了相應密碼子,合成插入部分胺基酸的肽鏈。又如Hb Grady α鏈第119與120間嵌入了α鏈第117~119三個胺基酸(苯丙-蘇-脯氨酸)。
(五)融合基因 減數分裂時,不同珠蛋白基因之間發生不等交換,合成融合鏈的異常血紅蛋白,如δ鏈和β鏈基因錯誤聯合,產生不等交換,形成融合基因δβ(Hb Lepore)和βδ(Hb反Lepore)。
(六)其它 由於α珠蛋白基因缺陷,使α鏈合成減少或缺如,過剩的β鏈與γ鏈形成四聚體,如β4-HbH,γ4Hb Barts;或由於β珠蛋白基因缺陷,βmRNA缺乏或轉錄、轉譯缺陷,使β鏈合成減少或缺如、導致HbA減少,而HbF、HbA2增高。上述珠蛋白肽鏈本身並無胺基酸順序的改變。
【血紅蛋白病的分子病理與溶血機理】
異常血紅蛋白病種類繁多,臨床症狀多樣化,但歸納其結構變異所導致功能異常,大致可分為以下數類:
1.因分子內部胺基酸替代所產生的異常血紅蛋白,血紅蛋白分子內部為非極性胺基酸,在血紅蛋白分子中構成血紅素與珠蛋白鏈的接觸,肽鏈螺鏇段間的接觸及血紅蛋白單體間的接觸,如被不同理化性質的胺基酸替代,會影響分子的構型和穩定性。此類異常血紅蛋白包括血紅蛋白M(HbM),不穩定血紅蛋白(UHb)和氧親和力改變的血紅蛋白。
(1)HbM:肽鏈中與血紅素鐵原子連線的組氨酸被酪氨酸所替代,最常見的是E7或F8的組氨酸為酪氨酸所替代,酪氨酸酚基上的氧與血紅素的鐵原子構成離子鍵,使鐵原子呈穩定的高鐵狀態,影響血紅蛋白的正常釋氧功能,使組織供氧不足,出現紫紺及紅細胞增多。高鐵血紅素並易與珠蛋白鏈分離,使血紅蛋白分子結構不穩定而發生溶血。
(2)UHb:肽鏈中與血紅素緊密結合的胺基酸發生替代或缺失,影響肽鏈的立體結構或減弱與血紅素的結合力,形成UHb分子。水易進入血紅蛋白袋內,使亞鐵血紅素氧化為高鐵血紅素;β第93位半胱氨酸的硫氫基被氧化,產生硫化物,形成硫化血紅蛋白,使珠蛋白鏈與血紅素分離。游離珠蛋白鏈在37℃即不穩定,四聚體易解離為單體,在紅細胞內聚集沉澱,形成包涵體,使細胞膜僵硬,通過微循環時往往導致膜部分喪失,最終變為球形紅細胞,在脾臟阻留而破壞。?蛋白種類很多,一般均對分子構型、功能和穩定性沒有明顯影響。HbE是β鏈第26位谷氨酸被賴氨酸替代。因谷、賴兩種胺基酸理化性質相同,其替代位置雖在α1β1接觸面上,但對血紅蛋白分子的穩定性和功能影響不大。這類異常血紅蛋白中少數可產生溶解度改變,如HbS和HbC均由於其分子外部形狀或電荷改變,缺氧時溶解度降低;HbS聚合為螺鏇狀體,扭曲成鐮刀形纖維;而HbC聚合為一種副結晶;兩者均使細胞膜變硬,難以通過微循環,喪失部份紅細胞膜,形成球形紅細胞,在脾竇內阻留溶破。
β-海洋性貧血患者,過剩的α肽鏈形成多聚體,引起紅細胞膜損害,致使大量幼紅細胞無效生成。α海洋性貧血,過剩的β及γ鏈形成HbH(β4)或HbBart(γ4)。HbH是一種不穩定血紅蛋白,HbH包涵體結合在紅細胞膜上,使膜對陽離子通透性發生改變,鉀鹽與水逐漸從紅細胞內滲透至細胞外。缺鉀紅細胞壽命縮短,易在單核/吞噬細胞系統破壞,導致溶血。Hb Bart對氧親和力增高,造成組織缺氧。
【診斷】
本病分布因地區、民族而異,故應詳細詢問患者籍貫、民族,臨床有無黃疸、貧血、肝脾腫大,生長發育遲緩或紫紺、紅細胞增多等;家系中有無同樣病史患者。實驗室檢查包括網織紅細胞計數、紅細胞壓積、周圍紅細胞形態及紅細胞脆性試驗,了解有無低色素、小細胞性貧血。如上述檢查提示有血紅蛋白病可能,應對患者及其家系作下列有關實驗室檢查,進一步確診。
我國北京、上海等大城市已建立先進的基因診斷技術,對多種遺傳血液病如血友病,α海洋性貧血、β-海洋性貧血、異常血紅蛋白病等成功地進行了基因診斷和產前基因診斷:
1.常用基因診斷方法為抽提全血、乾紙片血、羊水細胞、絨毛細胞DNA作DNA點雜交,適用於診斷基因缺失的遺傳病,如α海洋性貧血病人α珠蛋白基因不同程度的缺失。
2.限制性內切酶酶譜法,適用於診斷基因突變改變了限制酶切點或DNA缺失而改變酶解片段大小長短的遺傳病。
3.限制性片段多態性分析(RFLP),RFLP按孟德爾方式遺傳,如某種遺傳病基因與特異的RFLP緊密相連,即可將這一多態片段作為"遺傳標記",通過RFLP連鎖分析推測該家庭成員和胎兒是否攜帶遺傳病基因,RFLP連鎖分析適用於診斷任何一種單基因遺傳病。
4.寡核苷酸雜交是一種直接基因診斷技術,對於基因突變部位的鹼基序列已查明的遺傳病,均可以直接檢測和鑑定其突變的基因。
5.聚合酶鏈反應(PCR)DNA體外擴增,此種高效DNA分析技術可以直接通過PCR產物的電泳分析進行基因診斷,適用於診斷基因缺失或部分DNA缺失所致的遺傳病。
6.對非缺失型突變基因可結合限制酶切位點的改變,如與RFLP位點相連鎖,則可用限制酶消化PCR擴增產物,直接電泳分析,不需套用基因探針進行分子雜交,大大簡化實驗操作,使基因診斷可在半天內完成。
【治療說明】
目前尚無根治方法。對患者家系及本病高發地區,應做好血紅蛋白病普查,遺傳諮詢和婚前檢查。必要時進行產前診斷,做好優生工作,防止嚴重型血紅蛋白病患兒的出生。對重型患者可給予超量輸血,使用鐵螯合劑,減少含鐵血黃素沉著。脾功能亢進時可切除脾臟。至於對珠蛋白合成的調控,控制外源性珠蛋白基因在宿主細胞內正確有效的表達,目前尚在實驗研究階段,未能用於臨床,作為海洋性貧血的基因治療方法。
配圖
所屬分類
血液系統疾病現代醫學
