
簡介
另外,羧基不能被還原成醛基,要還原羧基必定是用很強的還原劑(LiAlH4),生成的醛會立即被還原。
此外由於羧基的特殊結構,使它還具有一定醛基(-CHO)的性質。
用新制氫氧化銅辨別醛基與羧基.現象:羧酸中藍色絮狀沉澱消失,變成藍色溶液,加熱不變化。反應原理是:酸鹼中和
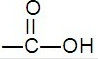
羧基
羧基會與羥基發生酯化反應—COOH+—OH=H2O+—COO—
檢驗試劑:飽和碳酸氫鈉溶液。
注意:羧基和酯基中的碳氧雙鍵一般不能發生加成反應,除非與強還原劑(LiAlH4等)反應。
官能團結構:
體例結構
 羧基脫水縮合
羧基脫水縮合羧基是有機化學中的基本酸基,所有的有機酸都可以叫羧酸,由一個碳原子、兩個氧原子和一個氫原子組成,化學式-COOH。如醋酸(CH3COOH)、檸檬酸都含有羧基,這些羧基與烴基直接連線的化合物,叫作羧酸。
羧基是由羰基和羥基組成的基團,它是羧酸的官能團,為羧基—COOH。
由羰基和羥基組成的一價原子團,叫做羧基。羧基的性質並非羰基和羥基的簡單加和。例如,羧基中的羰基在羥基的影響下變得很不活潑,不跟HCN、NaHSO3等親核試劑發生加成反應,而它的羥基比醇羥基容易離解,顯示弱酸性。在羧酸鹽的陰離子中,由於電子的離域作用,發生鍵的平均化。因此它的兩個碳氧鍵實際上是完全相等的。
此外由於羧基的特殊結構,使它還具有一定醛基(CHO-)的性質。
可以用新制氫氧化銅檢驗羧基的存在.現象:藍色絮狀沉澱消失,變成藍色溶液。反應原理是:氫氧化銅里的氫氧根被羧基氧化,使不可容的氫氧化銅變成可溶的氧化銅。
檢驗方法
可以用新制氫氧化銅檢驗羧基的存在.現象:藍色絮狀沉澱消失,變成藍色溶液即可。
HOOC--即是--COOH即羧基,--OH是羥基。
羧基的檢驗方法,即為檢驗酸的通性的方法(如使石蕊變紅等),檢驗羧基還可以用與醇類酯化的方法,但現象不一定很明顯。
驗證有機物中是否含有羧基一般只需加乙醇和濃硫酸,加熱。現象:會產生有香味的油狀物時即可。但最保險的方法還是用核磁共振。
測定含量
在織物的多元酸防皺整理中,經常需要測定羧基的含量,現將有關測定羧基的方法匯集如下。1.鉻黃法
鉻黃法的基本原理是基於氧化纖維素中的羧基能與某些重金屬(如鐵、鋁等)生成鹽類,通過複分解反應使沉澱在纖維上的金屬鹽呈現不同色澤,而正常纖維素無此反應。利用上述現象可用以鑑定羧基的存在及其含量的多少。鉻黃法使用的試劑是醋酸鉛和鉻酸鉀,反應如下:
Pb(Ac)2+R-(COOH)2→R-(COO)2Pb+2HAc
R-(COO)2Pb+K2CrO4→PbCrO4↓+R-(COOK)2
取試樣一塊,在50ml1%醋酸鉛溶液中處理5min。取出水洗後再浸入50ml1%鉻酸鉀溶液中處理5min。取出水洗,烘乾。試樣上含有羧基處即呈現黃色鉻酸鉛沉澱。
為求效果明顯,測定前,可將試樣用0.5%鹽酸溶液,以25:1浴比於室溫下處理40min,再用無離子(Ca2+,Mg2+)水抽濾洗滌到洗液中不含氯離子(用AgNO3檢查),晾乾。
2.拒染實驗法
利用氧化纖維素含有的羧基對直接染料的拒染性質,將棉纖維用直接染料染色。氧化纖維素不能染著或色澤很淺,而正常棉纖維可染得較深色澤。
將試樣一小塊投入300ml直接藍6B染浴(每升含染料5g)中,升溫到沸,染色5min。染色過程中試樣宜經常翻動。染後用70℃溫水洗淨,烘乾。觀察試樣色澤,氧化纖維素表現為拒染。
3.醋酸鈣法
氧化纖維素中羧基含量可用醋酸鈣法定量測定。氧化纖維素中的羧基能與醋酸鈣發生置換反應:
R-(COOH)2+(CH3COO)2Ca→R-(COO)Ca+2CH3COOH
生成的游離醋酸,可用氫氧化鈉標準溶液滴定。
試液配製
甲酚紅、百里酚藍混合指示劑:稱取0.008g百里酚藍溶於8ml乙醇中,加蒸餾水40ml。稱取0.004g甲酚紅,溶於4ml乙醇中。再加蒸餾水到20ml。然後將上述兩種溶液混合。
二乙基巴比妥酸和巴比妥鈉緩衝溶液:精確稱取巴比妥酸、巴比妥鈉各1g,分別用少量蒸餾水溶解(如不易溶解,稍加熱)。各放入50ml容量瓶中,用蒸餾水稀釋到刻度。混合液pH為8.3。
為使纖維素中的羧基以游離狀態存在,需先將試樣浸入0.5%鹽酸溶液中,浴比25:1,室溫放置40min,再用無離子水抽濾洗滌到織物不帶酸性[c(AgNO3)=0.1mol/L的硝酸銀溶液檢驗洗液到無白色AgCl沉澱為止。取出晾乾。再將試樣分成單紗,剪短,在大氣中平攤放置24h待用。
準確稱取試樣1g(精確到0.001g)兩份(平行試驗),分別放在兩個100ml碘量瓶中。用移液管吸取c(CaAc2)=0.1mol/L新鮮配製的醋酸鈣溶液50ml,加入到試樣中去。蓋塞,放置12-17h,並經常搖動,然後用移液管吸取25ml試液。為防止紗頭吸入,可用紗布包紮移液管吸入口。將試液放入100ml錐形瓶中,加甲酚紅及百里酚藍混合指示劑10滴,用c(NaOH)=0.01mol/L的氫氧化鈉標準溶液滴定,直至溶液色澤由黃色轉到紫玫瑰色。與事先準備好的標準溶液的色澤比較(取緩衝溶液25ml放入100ml錐形瓶中,加10滴指示劑),確定其終點,讀取耗用的c(NaOH)=0.01mol/L氫氧化鈉標準溶液毫升數V1。
平行做兩個空白試驗。稱取25ml空白試驗的醋酸鈣溶液,再用c(NaOH)=0.01mol/L氫氧化鈉標準溶液按上述方法滴定,讀取耗用的氫氧化鈉標準溶液毫升數V2。
計算
(V1-V2)×c(NaOH)×50
羧基含量=——————————————×100%
W
式中W為纖維素試樣重,必須換算成絕對乾燥重代入。
4.亞甲基藍吸收值
亞甲基藍為一個鹽基性染料,它不能染著纖維素纖維,但當纖維素被氧化生成部分羧基後,基於離子交換作用,能吸附鹽基性染料而被染著。亞甲基藍以MB+Cl-表示。它和纖維素中羧基的作用為:
R-COOH+MB+Cl-?R-COOMB+H++Cl-
因此,纖維素羧基的含量可以定量的用吸附亞甲基藍的數量表示。亞甲基藍值是指100克絕對乾燥纖維吸附亞甲基藍的毫摩爾數。
由於上染是一個可逆過程,纖維上染料的吸附平衡受染料濃度和H+濃度的影響,此外染液中的Na+也能和羧基作用而與亞甲基藍“競染”,從而影響了羧基對染料的吸附。因此,測定亞甲基藍值時必須規定嚴格的條件,即規定了始染液染料濃度為c(MB+Cl-)=0.40mmol/L,染液的pH=8。又由於纖維素羧基吸附亞甲基藍的量與亞甲基藍濃度之間並不呈線性關係,因此還規定一個染色平衡時的染料濃度,即規定在染色平衡時纖維素所吸附的染料量為始染時染料量的一半(50%吸盡率),並規定在此條件下Na+與亞甲基藍染料的濃度比應為4:1。
主要儀器和化學品
分光光度計
亞甲基藍鹽酸鹽(分子量320)、二乙基巴比妥酸、氫氧化鈉。
染液製備
將精確稱重的1.28g亞甲基藍[n(MB+Cl-)=4mmol]置於1000ml量筒內。加入c(NaOH)=0.1mol/L氫氧化鈉溶液80ml和2.30g二乙基巴比妥酸,將上述染化料充分溶解後,加蒸餾水到1L。
精確量取100ml上述溶液置於1L容量瓶中,加蒸餾水到1L,配製成每升含亞甲基藍n(MB+Cl-)=0.4mmol、氫氧化鈉n(NaOH)=0.8mmol以及pH=8的溶液。
制定工作曲線
精確量取c(MB+Cl-)=0.4mmol/L亞甲基藍溶液10mL、25mL、50mL和75mL各1份於100mL容量瓶中,加蒸餾水到100mL。其濃度c(MB+Cl-)依次為0.04mmol/L、0.10mmol/L、0.20mmol/L和0.30mmol/L。然後每份各取10mL,加上未稀釋的染液10mL,各置於100mL容量瓶中,加c(HCl)=0.1mol/L鹽酸溶液到刻度。其濃度依次為0.004mmol/L、0.010mmol/L、0.020mmol/L、0.030mmol/L和0.040mmol/L。
在分光光度計上選定最大吸收波長,用1cm玻璃皿測出各個染液的吸光度,作出染液濃度和吸光度的工作曲線。
精確稱取0.1-2.0g纖維素試樣,重量應按其對染料的規定吸附量(50%吸盡率)選定。試樣應先經乾燥並用五氧化磷處理後稱重,或用另一塊相同的試樣在110℃烘到恆重計算含水率後,在測試用試樣的重量中扣除含水率,以求得其絕對乾燥重。
將試樣剪成小塊置於100mL燒杯中,加入100mLc(MB+Cl-)=0.40mmol/L亞甲基藍溶液,保持經常搖動,在室溫下染色2h。
染色完畢後,將染液及試樣經2#玻璃砂芯漏斗過濾,準確吸取10mL放入100mL容量瓶中,用c(HCl)=0.1mol/L鹽酸溶液被稀釋到刻度,在分光光度計上測定其吸光度,求出濃度值。
在測定完畢後應重新校正一次c(MB+Cl-)=0.4mmol/L原始亞甲基藍染液的吸光度值。按規定,染後的染液濃度應是始染濃度的50%,因此,始染液濃度為c(MB+Cl-)=0.4mmol/L,染後平衡濃度應為c(MB+Cl-)=0.02mmol/L。如測定濃度大於此數,應增加試樣重量,反之應減少試樣重量,因此本試驗需經多次測量及測定才能調整到適當的試樣重量。
計算
每升染液中纖維上亞甲基藍吸附量=(x-y)(mmol)
(x-y)
每100mL染液中纖維上亞甲基藍吸附量=—————(mmol)
10
(x-y)100
亞甲基藍值=—————×———(mmol/100g纖維)
10W
式中:x——始染染液濃度(mmol/L);
y——染後染液濃度(mmol/L);
W——試樣重(g)。
5.醋酸鈣法(之二)
試樣用0.5%HCl浸泡40min,再用去離子水洗滌到無Cl-,晾乾,乾燥,並在乾燥器中保存。準確稱取兩份各1g的試樣,用新鮮配製的0.1M醋酸鈣溶液浸漬於250ml碘量瓶中。放置12-17h,並經常搖動。吸取10ml試液於250ml錐形瓶中,以甲酚紅及百里酚藍為混合指示劑,用0.1mol/L的NaOH標準溶液滴定,至溶液色澤由黃色轉到紫玫瑰色。記下NaOH的起始值。計算含量:
羧基含量(mol/g)=2×(V-V0)×c×50/1000W
V:整理後試樣消耗NaOH體積ml;
V0:空白試樣消耗NaOH體積ml;
W:試樣質量g;
C:NaOH濃度mol/L
保護方法
 複雜的芳香族多羥基羧基的物質
複雜的芳香族多羥基羧基的物質1.酯化法保護羧基:甲酯和乙酯
甲酯和乙酯作為羧酸的保護基對一系列合成操作十分適用。例如,以酯的形式進行的烷基化反應和各種縮合反應,隨後酯基在酸或鹼的催化下水解除去,偶爾酯基也可用熱解反應消去。但簡單的烷基酯作為羧酸的保護基在有些情況下並不適用,其原因往往是由於最後需用皂化反應來除去酯基。因此,實際上在合成中常甲基和乙基的衍生物取而代之。甲基的衍生物主要是苄基類型,可用溫和條件下的酸處理或氫解脫除。乙基衍生物主要是β,β,β2三氯乙基等
2.酯化法保護羧基:叔丁酯
叔丁酯不能氫解,在常規條件下也不被氨解及鹼催化水解,但叔丁基在溫和的酸性條件下可以異丁烯的形式裂去。此性質使叔丁基在那些不能進行鹼皂化的情況下特別吸引人,例如:用於酮、β2酮酯、α,β不飽和酮和對鹼敏感的α2酮醇以及肽的合成。在青黴素的合成中,可選擇性地裂開叔丁酯以便形成β2內醯胺;在菌黴素的合成中和在容易還原的酮的製備中,都可用叔丁基來保護羧基。四氫吡喃酸具有和叔丁酯相似的對酸的不穩定性,這一保護基也類似地用於丙二酸酯類型的酮和酮酯的合成中。
3.酯化法保護羧基:苄基、取代苄基及二苯甲基酯類
這類酯保護基的特點在於它們能很快地被氫解除去。在青黴素合成中,苄酯不被溫和的酯水解條件破壞,最後需由氫解除去苄酯;在谷醯胺和天門冬醯胺的合成中,以及在L2谷氨酸和L2天門冬氨酸酯的製備中,苄酯的性質都能典型地顯示出來。Bowman和Ames將苄基酯用在活性酯(有α2活潑氫)的烷基化或醯基化中,此法曾出色地完成脂肪酸、酮、二酮和α2醇酮的合成。芳環上或次甲基上有取代基的苄基在用酸性試劑脫去時,其敏感性可有大幅度的改變。Stewevr在酯肽類合成中利用了亞甲苄酯易於催化脫去的優點,用其代替叔丁酯。苄酯和對硝基苄酯也可作為羧基的保護基,一個典型的例子就是其在氨基的醯化衍生物合成中的套用。在苯酯和縮酚酸的合成中,二苯甲酯具有相似的作用,但二苯甲酯在酸存在條件下的溶劑化分解太快,因此在酸性條件下不易作羧基保護基。總之,這類酯是一種有價值的保護基,其製備可用經典的方法及前述的反應製備。
4.用醯胺和醯肼來保護羧基
在有限的範圍內人們採用醯胺和醯肼的形式保護羧基,從其解脫方式的角度補充了酯類保護作用的不足。醯胺和醯肼對解脫酯類的溫和鹼性水解條件穩定,但酯類對能有效脫解醯胺的亞硝酯和用於裂解醯肼的氧化劑又均穩定,二者可以互補。
製備醯胺和醯肼的經典方法是以酯或醯氯分別與胺或肼作用製備,也可直接從酸制
得。醯肼已被用於抗菌素和肽的合成,在肽的合成中它們可被亞硝酸轉化為疊氮化物,使得縮合反應容易發生。
5.酯的保護
酯和內酯的保護可視為羧基的間接保護,而且酯須有α2活潑氫,否則反應很複雜。酯在引進保護基後,可在很多條件下保持穩定,如HOAc/H2O/THF(25℃,1h),KOH/MeOH(25℃,12h),LiAlH4/Et2O(25℃,3h),CH3Li/Et2O(25℃,2h)等。可用汞鹽或三氟化硼脫去脂保護基。
綜上所述,保護羧基的方法雖然不多,但作為保護基的酯的種類卻不少,且各有特色。近年來有關羧基保護的研究主要在肽、胺基酸、抗菌素等的合成方面,且套用曰見廣泛。
