發病機制
 健康
健康SDS的病因不明目前涉及的有脂質過氧化損傷酶代謝異常慢病毒感染神經元凋亡少突膠質細胞胞質內包涵體等,導致的進行性神經系統多系統變性。
病理改變病理改變決定臨床表現SDS病理上主要的改變是自主神經中樞即脊髓中間外側柱節前交感神經元明顯減少有瀰漫性變性,骶2~4節段Onuf核細胞完全消失,交感節和節後纖維也變性。
 健康
健康此外中樞神經系統其他部位如錐體外系小腦錐體系和前角細胞也可受累,特別是腦橋黑質、小腦蒲肯野細胞、藍斑、尾狀核、殼核下橄欖核、迷走神經背核等廣泛神經細胞變性、脫失,伴有神經膠質增生;65%以上病例橄欖體腦橋小腦及黑質紋狀體系統受累前角細胞受累不太常見。
上述神經結構原發性變性,與反覆直立性低血壓無關,是有關神經結構多巴胺和腎上腺的耗竭。膽鹼能神經功能研究結果表明,它是副交感節後膽鹼能與腎上腺素能障礙。
臨床表現
 夏伊-德雷格綜合症
夏伊-德雷格綜合症本病起病隱襲,可從數月至數年長者可達10年以上由中老年起病,發病年齡平均55歲。約65%為男性無家族史病程為進展性,自主神經症狀常首先出現,數月或數年後方見軀體神經症狀。也有少數患者軀體神經症狀早於自主神經症狀。
直立性低血壓臨床早期表現為突然起立或站立過久時出現頭暈患者常有視物模糊和易疲勞感,患者常因直立性暈厥就診由於自主神經的廣泛變性,壓力感受器反射弧被阻斷,直立時不能產生反射性心率增加和周圍小動脈收縮,而出現直立性低血壓直立性低血壓是最常見症狀,臥位血壓正常,也有高於正常者,直立時血壓顯著下降[收縮壓下降超過4.0~6.7kPa(30~50mmHg)],立位與臥位血壓在2min內常常相差4.0/2.67kPa(30/20mmHg)由於交感神經張力低下使患者站立時脈率無改變沒有面色蒼白,噁心嘔吐多汗等症狀。據統計在直立性低血壓患者中占11%。
其他自主神經功能障礙如膀胱、直腸括約肌障礙引起排尿、排便障礙包括尿頻尿急尿瀦留或失禁腹瀉和便秘交替性慾障礙可以在其他神經系統症狀出現前數年即有出汗異常可先為多汗後為少汗或皮膚乾燥、無汗另外可有少見的皮溫異常霍納綜合徵、虹膜萎縮等。本病多從骶髓開始逐漸向上發展,故出現症狀順序是首先出現陽痿性慾減退、排尿障礙,後出現頭昏眼花、暈厥、直立性低血壓等自主神經功能障礙症狀,最後出現小腦性共濟失調、腦幹損害症狀及錐體束等症狀。
小腦功能障礙如意向性震顫,肢體、軀幹共濟失調,輪替動作差眼震言語不清構音障礙。直立性低血壓自主神經功能障礙及小腦症狀三主征是臨床核心症狀。也可合併由於錐體外系、基底核或脊髓運動神經元變性引起的軀體神經異常晚期可有錐體束損害如肌張力增高腱反射亢進病理征陽性等;在肢體遠端可有肌萎縮和肌束顫動肌電圖顯示前角細胞變性性改變;也可由於黑質受累表現帕金森綜合徵。也可有腦神經癱瘓等。
夜間發生喉哮鳴(1aryngealstridor)是由於喉外展肌力弱造成的,並可伴有呼吸暫停相當一部分患者發生吞咽困難。晚期可因自主神經功能極度衰竭可在睡眠中呼吸暫停死亡。在疾病後期常見情緒不穩、抑鬱,晚期可表現精神衰退甚至痴呆、臥床不起。隨病情發展出現多種的症狀體徵可以是本病表現,也可以看作本病併發症(參見臨床表現)。另外應注意繼發的肺部感染尿路感染等
主要依據
 夏伊-德雷格綜合症
夏伊-德雷格綜合症(1)散發性成年潛隱起病的進行性自主神經功能障礙臨床表現為直立性低血壓,立位收縮壓較臥位下降4~6.67kPa(30~50mmHg),舒張壓較臥位下降2.67kPa,而心率變化不大,陽痿或閉經,發汗障礙排尿功能障礙及瞳孔改變等。
(2)出現帕金森綜合徵。
(3)出現小腦征。
(4)出現錐體束征。
(5)排除其他疾病。
以上5項中(1)必備,(2)、(3)兩項具備一項即可(4)、(5)作為參考。
2.臨床診斷可分為3型:
(1)進行性自主神經功能障礙。
(2)進行性自主神經功能障礙伴帕金森綜合徵。
(3)進行性自主神經功能障礙伴小腦征。
鑑別診斷
 夏伊-德雷格綜合症
夏伊-德雷格綜合症SDS患者的暈厥與血管迷走性暈厥有所不同,後者常有多汗面色蒼白和噁心等表現與OPCA、SND的主要鑑別點為:夏伊-德雷格綜合徵早期出現直立性低血壓陽痿、尿便功能障礙等自主神經症狀。需根據小腦錐體外系自主神經症狀的側重予以判斷。
檢查輔助
 夏伊-德雷格綜合症
夏伊-德雷格綜合症腦脊液檢查一般無異常改變。
腦電圖與誘發電位部分病例有輕度改變無特異性。
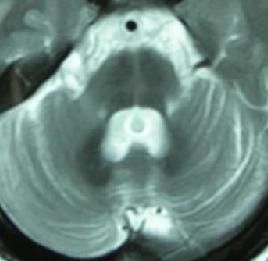
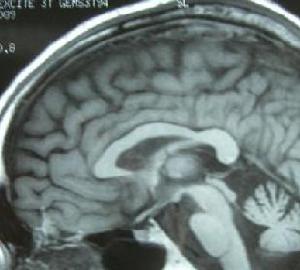
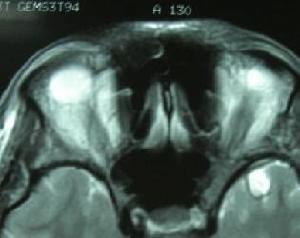
MRI或CT檢查顯示腦幹小腦有不同程度的體積縮小,腦池、腦室有不同程度的擴大及大腦皮質腦溝增寬等也可表現為正常。
1986年DragerBehram同時報導了一組SDS的MRI表現。發現這些患者的殼核均呈短T2信號。以後Mario等認為殼核短T2信號對SDS的診斷具有診斷價值但對於成因觀點不一有作者認為源自一種順磁性物質——鐵元素,即可能與在此部位過度沉積的鐵有關細胞內鐵過多可加速去相位過程,縮短T2值在MRIT2加權像呈低信號。鐵元素沉積於殼核治療:
無特效治療,主要是對症治療,1.直立性低血壓的治療見MSAMatsubara等套用氟氫可的松(9-α氟氫可的松)及左鏇-蘇-34-雙氫苯基絲氨酸(DOPS)治療SDSFC每天晨服0.1~0.2mg;左鏇-蘇-3,4-雙氫苯基絲氨酸(DOPS)先每天晨服100mg,2周內增至200mg3次/d結果表明,氟氫可的松(FC)消除起立時血壓的明顯下降還有減輕rCBF下降的趨勢;而左鏇-蘇-3,4-雙氫苯基絲氨酸(DOPS)能減輕起立時舒張壓和rCBF的明顯下降其副作用少可替代氟氫可的松(FC)用於SDS的治療。
帕金森綜合徵伴有帕金森綜合徵的SDS患者治療更加困難,因為多巴胺能藥物常使已經很明顯的直立性低血壓症狀更加惡化,而且多巴胺能藥物常對MSA患者的帕金森綜合徵無效。
其他神經源性臥位高血壓常見,通常不需要治療抬高床頭15°~20°即可控制也可使用糖皮質激素及免疫抑制藥(環磷醯胺、硫唑嘌呤)治療。中藥可用生脈散治療生脈散能益氣養陰生津。血管內皮細胞對鐵的攝取和轉運過程發生障礙有關。
可能與此部位的毛細 夏伊-德雷格綜合徵(Shy-Drgersyndrome,SDS)是以植物神經受損為主要表現的,多系統萎縮(multiplesystematrophy,MSA)疾病中的一種類型,MSA是一種原因不明的中樞神經系統多部位萎縮變性疾病。本組3例SDS病人均為1994~1998年門診及病房確診的患者,並對其交感皮膚反應(sympatheticskinresponse,SSR)檢查所見進行了分析。
實例說明
 夏伊-德雷格綜合症
夏伊-德雷格綜合症結果: 3例SDS患者均為隱襲性發病,初次就診距發病時間為2~6年,平均4年。3例均有頭暈、尿頻症狀,其中2例有尿瀦留、尿失禁、陽萎和便秘,1例並有暈厥。3例均有直立性低血壓,臥、立位收縮壓下降45~70mmHg(1mmHg=0.133kPa),平均下降58mmHg,舒張壓下降15~25mmHg,平均下降20mmHg。3例均有行走不穩,共濟失調和龍貝格征陽性及下肢單側或雙側巴賓斯基征陽性和查多克征陽性,植物神經檢查,2例患者胸3、4以下皮膚乾燥、無汗,雙下肢皮膚劃痕試驗白色明顯延長,1例患者手足及軀幹皮膚乾燥、皸裂,指甲粗糙。全部行頭顱MRI掃描,2例有腦幹及小腦萎縮,1例行P300檢查潛伏期延長P3b412ms,3例患者行SSR下肢檢查,1例SSR未測出,另2例SSR波幅低限(0.1μV,0.12μV)結果全部異常。
目前國內對SDS的SSR檢測尚無報導。SSR是臨床肌電圖實驗室中一種新的、用來評估部分周圍神經系統——小的無髓C纖維的方法[2],由Shahami等1984年首次報導,SSR檢測與腓腸神經活檢病理對比結果表明,SSR是評定無髓軸索疾病皮膚交感纖維的特定檢查方法。此後,Baser等[3]在MSA患者泌汗減少方面進行了研究;Ravits等[4]又提出了SSR結合瓦薩瓦比率和深吸氣異常心率變化診斷MSA準確率可達91%,男性患者SSR檢測更有價值,女性患者由於分娩陰部神經損傷可造成較高的假陽性率。Bordet等[5]也通過實驗證實SSR在MSA和特發性帕金森病患者中有鑑別診斷意義,可排除誤診為特發性帕金森病的MSA患者。
