
 傳統的配位化合物
傳統的配位化合物配合物是化合物中較大的一個子類別,廣泛套用於日常生活、工業生產及生命科學中,近些年來的發展尤其迅速。它不僅與無機化合物、有機金屬化合物相關連,並且與現今化學前沿的原子簇化學、配位催化及分子生物學都有很大的重疊。
概述
討論經典配位化合物時,常會提到以下的術語:
1.配位鍵、配位共價鍵:配位化合物中存在的化學鍵,由一個原子提供成鍵的兩個電子,作為電子給予體,另一個2.成鍵原子則作為電子接受體。參見酸鹼反應和路易斯酸鹼理論。
3.配位單元:化合物含有配位鍵的一部分,可以是分子或離子。
4.配離子:含有配位鍵的離子,可以是陽離子或陰離子。
5.內界、外界:內界指配位單元,外界與內界相對。
6.配體、配位體、配位基:提供電子對的分子或離子。
7.配位原子:配體中,提供電子對的原子。
8.中心原子、金屬原子:一般指接受電子對的原子。
9.配位數:中心原子周圍的配位原子個數。
10.螯合物:含有螯合配體的配合物。
此外,含有多箇中心原子的配合物稱為多核配合物,連線兩個中心原子的配體稱為橋聯配體,以羥基橋聯的稱為羥聯,以氧基橋聯的稱為氧聯。
歷史
人們很早就開始接觸配位化合物,當時大多用作日常生活用途,原料也基本上是由天然取得的,比如殺菌劑膽礬和用作染料的普魯士藍。最早對配合物的研究開始於1798年。法國化學家塔薩厄爾首次用二價鈷鹽、氯化銨與氨水製備出CoCl3.6NH3,並發現鉻、鎳、銅、鉑等金屬以及Cl-、H2O、CN-、CO和C2H4也都可以生成類似的化合物。當時並無法解釋這些化合物的成鍵及性質,所進行的大部分實驗也只局限於配合物顏色差異的觀察、水溶液可被銀離子沉澱的摩爾數以及電導的測定。對於這些配合物中的成鍵情況,當時比較盛行的說法借用了有機化學的思想,認為這類分子為鏈狀,只有末端的鹵離子可以離解出來,而被銀離子沉澱。然而這種說法很牽強,不能說明的事實很多。
1893年,瑞士化學家維爾納總結了前人的理論,首次提出了現代的配位鍵、配位數和配位化合物結構等一系列基本概念,成功解釋了很多配合物的電導性質、異構現象及磁性。自此,配位化學才有了本質上的發展。維爾納也被稱為“配位化學之父”,並因此獲得了1913年的諾貝爾化學獎。
1923年,英國化學家西季威克提出“有效原子序數”法則(EAN),提示了中心原子的電子數與它的配位數之間的關係。很多配合物,尤其是羰基配合物,都是符合該法則的,但也有很多不符合的例子。雖然這個法則只是部分反映了配合物形成的實質,但其思想卻也推動了配位化學的發展。
現代的配位化學不再拘泥於電子對的施受關係,而是很大程度上藉助於分子軌道理論的發展,開始研究新類型配合物如夾心配合物和簇合物。其中一個典型的例子便是蔡氏鹽—K[Pt(C2H4)Cl2]。雖然該化合物早在1827年便已經製得,但直到1950年才研究清楚其中的反饋π鍵性質。
分類
配位化合物可分成傳統配位化合物及有機金屬化合物。
1.傳統配位化合物由一個以上的配離子(也叫離子複合物)形成,配位鍵中的電子“幾乎”全部由配體提供。典型的配體包括H2O、NH3、Cl−、CN−和en-。
例子:[Co(EDTA)]−、[Co(NH3)6]Cl3、[Fe(C2O4)3]K3和[Cr(H2O)6]Cl3。
2.有機金屬化合物指含有金屬-碳化學鍵的化合物,配體為有機基團(如烯烴、炔烴、烷基、芳香環)或性質類似的化學品,如膦、氫負離子、一氧化碳。
例子:(C5H5)Fe(CO)2CH3、Fe(CO)5、Cp2TiMe2。
與配位化學有交蓋的化學分支如:
1.生物無機化學——其中的配合物配體存在於自然界中,常為胺基酸側鏈和輔酶,如卟啉。例子包括血紅素。
2.原子簇化學——用金屬原子作配體,如Ru3(CO)12。
結構
構型
配位化合物的構型由配位數所決定,也就是化合物中心原子周圍的配位原子個數。配位數與金屬離子和配體的半徑、電荷數和電子構型有關,一般在2-9之間,鑭系元素和錒系元素的配合物中常會出現10以上的配位數。
把圍繞中心原子的配位原子看作點,以線連線各點,就得到配位多面體。配位數與配合物構型的關係列在下表:
| 配位數 | 構型 | 圖形 | 3D | 實例 |
| 2 | 直線型 D∞h | 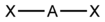 |  | HgCl2、Ag(NH3)2+、[Au(CN)2]- |
| 3 | 平面三角形 D3h | 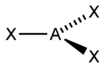 | 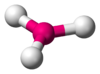 | HgI3-、Pt(PPh3)3、Fe[N(Si(CH3)3)2]3 |
| 4 | 四面體 Td | 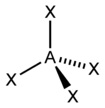 | 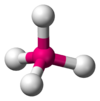 | Ni(CO)4、MnO4-、SnCl4 |
| 平面正方形 D4h | 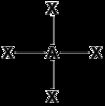 | 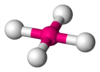 | cis-Pt(NH3)2Cl2、PtCl42-、Ni(CN)42- | |
| 5 | 三角雙錐 D3h | 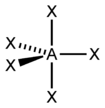 | 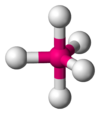 | Fe(CO)5、CdCl53- |
| 四方錐 C4v | 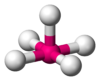 | [InCl5]2-、SbF52- | ||
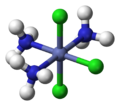
| 6 | 八面體 |  | 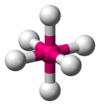 | [Ti(H2O)6]3+、[Co(en)3]3+、[Cu(NH3)6]2+ |
| 7 | 五角雙錐 D5h | 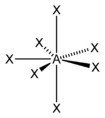 | 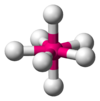 | [ZrF7]2-、[UO2F5]3- |
五配位中,常常涉及到三角雙錐和四方錐兩種構型的互變,因此,很大一部分五配位化合物的結構是介於這兩個結構之間的一種中間結構。六配位的化合物除極其常見的八面體外,也有可能是三角稜柱結構,例如單核配合物[Re(S2C2Ph2)3]即屬於這一類。七配位中,配合物還可能是單帽八面體或單帽三角稜柱體結構。
更高配位數的化合物中,八配位的可以是四方反稜柱體、十二面體、立方體、雙帽三角稜柱體或六角雙錐結構;九配位的可以是三帽三角稜柱體或單帽四方反稜柱體結構;十配位的可以是雙帽四方反稜柱體或雙帽十二面體結構;十一配位的化合物很少,可能是單帽五角稜柱體或單帽五角反稜柱體;十二配位的如[Ce(NO3)6]3-,為理想的二十面體;十四配位的為雙帽六角反稜柱體。再高的配位數非常罕見,如最近研究的PbHe152+,該離子中鉛的配位數至少為15。
以上只是配合物構型的理想情況。實際中的配合物結構常會發生畸變,原因可能是位阻效應、電子效應(參見姜-泰勒效應)或配體種類的緣故等。
異構現象
異構現象是配合物具有的重要性質之一。它不僅影響配合物的物理和化學性質,而且與其穩定性、反應性和生物活性也有密切關係。重要的配合物異構現象包括立體異構和結構異構。
立體異構
立體異構是化學式和原子排列次序都相同,僅原子在空間排列不同的異構現象。立體異構主要分為幾何異構和光學異構。
幾何異構
幾何異構是組成相同的配合物的不同配體在空間幾何排列不同而致的異構現象,主要出現在配位數為4的平面正方形和配位數為6的八面體結構中,以順式-反式異構體與面式-經式異構體的形式存在。
從空間關係上考慮,順式(cis-)是指相同的配體處於鄰位,反式(trans-)是指相同的配體處於對位。八面體[MA3B3]的兩種異構體中,面式(fac-)或順-順式指3個A和3個B各占八面體的三角面的頂點,經式(mer-)或順-反式是指3個A和3個B在八面體外接球的子午線上並列。見下圖:
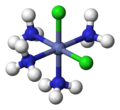 | 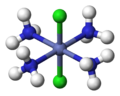 | 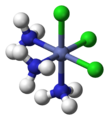 |  |
| cis-[CoCl2(NH3)4]+ | trans-[CoCl2(NH3)4]+ | fac-[CoCl3(NH3)3] | mer-[CoCl3(NH3)3] |
不對稱雙齒配體的平面正方形配合物[M(AB)2]也有可能有幾何異構現象,結構類似於上面的順鉑,見下圖:
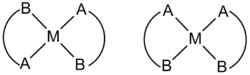 | 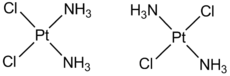 |
| 順鉑(左)與其反式異構體具有截然不同的抗癌活性 |
多核配合物也有幾何異構現象。例如,Pt(II)的雙核配合物[Pt2(PPr3)2(SEt)2Cl2]的順反異構體都已製得,且室溫下其苯溶液都是穩定的。但反式在熱的或冷的苯溶液中加入痕量三丙基膦作催化劑就能完全轉變為順式。
光學異構
光學異構是立體異構的另一種形式,兩種光學異構體會使平面偏振光發生等量但不同方向的偏轉,因此又稱鏇光異構或對映異構。大多數配合物在溶液中都會逐漸失去鏇光性,這一過程稱為消鏇作用。根據具體情況的不同,消鏇機理可能是分子間或分子內的。
最簡單的配合物光學異構體為四面體型,中心原子與四個不同的基團相連,分子不能與鏡像重合。例如[BeII(C6H5COCHCOCH3)2]。而對於八面體構型的配合物而言,光學異構主要發生在以下幾種情況下:
1.[M(AA)3]型,如三-(草酸根)合鉻(III)、[Co{(OH}2Co(NH3)4}3]Cl6(第一個製得的具有鏇光性且不含碳的化合物—Hexol)。
2.[M(AA)2X2]型,如[Rh(en)2Cl2]+。
3.[M(AB)3]型,如[Co(gly)3]。
4.[M(AA)B2X2]型,如[Co(en)(NH3)2Cl2]+。
5.涉及多齒配體,如[Co(edta)]-。
 |  | ![配合物Λ-cis-[CoCl2(en)2]](/img/d/77f/nBnauM3X0ETN4gTM0AzM1ITMyITM1YTOxIjMwADMwAzMxAzLwMzL3QzLt92YucmbvRWdo5Cd0FmLxE2LvoDc0RHa.jpg) | ![配合物Δ-cis-[CoCl2(en)2]](/img/1/bab/nBnauM3XxMTM0cDM5AzM1ITMyITM1YTOxIjMwADMwAzMxAzLwMzL4QzLt92YucmbvRWdo5Cd0FmLxE2LvoDc0RHa.jpg) |
| Λ-[Fe(ox)3]3− | Δ-[Fe(ox)3]3− | Λ-cis-[CoCl2(en)2]+ | Δ-cis-[CoCl2(en)2]+ |
結構異構
結構異構是化學式相同,但原子排列次序不同的異構體,主要可分為以下幾類:
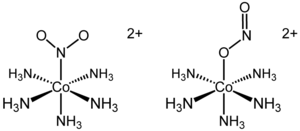 [Co(NH3)5(NO2)]2+的兩種鍵合異構體。
[Co(NH3)5(NO2)]2+的兩種鍵合異構體。構型異構:配合物可以採取一種以上的構型。比如[NiCl2(Ph2PCH2Ph)2]可分別呈四面體和平面四邊形構型。常見的構型異構有五配位化合物三角雙錐和四方錐構型之間的異構,以及八配位化合物十二面體和四方反稜柱體構型之間的異構。
配位體異構:互為同分異構體的配體所形成的類似配合物,如1,3-二氨基丙烷與1,2-二氨基丙烷分別形成的鈷配合物[Co(H2N-CH2-CH2-CH2-NH2)Cl2]、[Co(H2N-CH2-CH(-NH2)-CH3)Cl2]。
離子異構:配合物有相同分子式但不同的配位陰離子,因此水溶液中產生的離子不同,如[Co(NH3)5SO4]Br和[Co(NH3)5Br]SO4。
溶劑合異構:配合物中水所處的位置不同,有內界與外界的差異,例如[Co(H2O)6]Cl3和[Cr(H2O)5Cl]Cl.H2O。
配位異構:陽離子和陰離子都是配離子,且配體可以互相交換成分。例子有:[Co(NH3)6][Cr(CN)6]和[Cr(NH3)6][Co(CN)6]、[Cr(NH3)6][Cr(SCN)6]和[Cr(SCN)2(NH3)4][Cr(SCN)4(NH3)2],以及[PtII(NH3)4][PtIVCl6]和PtIV(NH3)4Cl2][PtIICl4]。
聚合異構:是配位異構的一種,用以表示配合物相對分子質量上的倍數關係,與聚合反應中的“聚合”並不類同。例如,[Co(NH3)6][Co(NO2)6]可看作[Co(NH3)3(NO2)3]的二聚體。
化學鍵理論
配位化合物的化學鍵理論,主要研究中心原子與配體之間結合力的本性,用以說明配合物的物理及化學性質,如磁性、穩定性、反應性、配位數與幾何構型等。配合物的理論起始於靜電理論。而後西季威克與鮑林提出配位共價模型,也就是套用配合物中的價鍵理論,統治了這一領域二十餘年,可以較好地解釋配位數、幾何構型、磁性等一些性質,但對配合物的顏色和光譜卻無能為力。
價鍵理論認為,配體提供的孤對電子進入了中心離子的空原子軌道,使得配體與中心離子共享這兩個電子。配位鍵的形成經歷了三個過程:(激發)、雜化和成鍵,其中雜化也稱軌道雜化,是能量相近的原子軌道線性組合成為等數量且能量簡併雜化軌道的過程。由此還可衍生出外軌/內軌型配合物的概念,從而通過判斷配合物的電子構型及雜化類型,就可以得出配合物的磁性、氧化還原反應性質以及幾何構型。對於很多經典配合物來說,價鍵理論得出的結果還是比較貼近事實的。
除了價鍵理論之外,而後發展的晶體場理論與配位場理論也是比較重要的配合物理論。
晶體場理論將配體看作點電荷,並將配位鍵當作離子鍵處理,可看作是靜電理論的延伸。並且,它以不同幾何構型中,配體對不同空間取向的d軌道的作用作為切入點,得出不同取向d軌道會發生能級分裂,並建立起分裂能及晶體場穩定化能的概念,以推測配合物的電子組態及穩定性。晶體場理論可以很好地解釋配合物的顏色、熱力學性質和配合物畸變等現象,但不能合理解釋配體的光譜化學序列,也不能很好地套用於特殊高/低價配合物、夾心配合物、羰基配合物和烯烴配合物。
配位場理論結合了分子軌道理論與晶體場理論。它在理論上更加嚴謹,然而定量計算則很困難,計算過程中不得不引進近似處理,因而也只能得到近似的結果。
各種反應
配位反應
配位反應可視同路易斯酸鹼理論中的酸鹼反應:金屬離子為酸提供空軌道,配體提供電子對為鹼,過渡金屬與配體的反應常伴隨著顏色的變化。例如,將HCl加入[Cu(NH3)4]2+依序產生[Cu(H2O)4]2+(淡藍色)、[CuCl(H2O)3]+、[CuCl2(H2O)2]、[CuCl3(H2O)]-、[CuCl4]2-、[Cu(NH3)4]2+(深藍色);再如,將過量的氨水加入[Cu(H2O)4]2+顏色立即由淡藍色轉變成深藍色:
[Cu(H2O)4]2+(淡藍)+4NH3→[Cu(NH3)4]2+(深藍)+4H2O
配體交換反應
配位化合物中的配體可被其它配體所取代,稱為配體交換反應,一般反應機理為親核取代反應。以八面體配合物為例,此類反應的通式為:
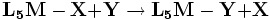 反應
反應這類配體交換反應的速率相差很大,有些反應在10-10秒內就可完成,而有的反應則需要數月。對於活性的差異有一個人為的規定,認為在濃度約為0.1M,溫度25°C時,半衰期
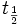 反應
反應價鍵理論和配位場理論都對這類反應的速率差異做了解釋,一般存在以下的規律:
1.中心金屬離子電荷的增加,會使反應速率降低;
2.中心離子為d0、d1、d2、d9、d10構型,低自鏇d5、d6、d7構型和高自鏇d4構型的配合物,對於配體交換反應都是活性的;
3.中心離子為d3、d8構型,或低自鏇的d4、d5、d6構型時,對於配體交換反應是惰性的。
此外,反應速率還與溶劑、配體的種類和排列有很大關係。
氧化還原反應
配位化合物的氧化還原反應包含兩種類型,一種是中心原子與配體之間的氧化還原反應,另一種則是兩個配合物之間的氧化還原反應。後者又可分為兩類:
1.電子轉移機理、外層反應機理:兩個反應物的第一配位層都保持不變。反應速率主要與反應物的結構與電子自鏇態有關,含有π共軛體系配體,如聯吡啶、CN-的配合物反應速率往往較快。此外,橋聯配體也可傳遞電子,但一般不如直接的電子轉移反應有效。
2.橋式機理、內層反應機理:兩個金屬原子同時連線在一個橋聯配體上,組成過渡態。
反應是以外層機理進行,還是以內層機理進行,與配合物的結構有關。對配體交換反應呈惰性、沒有橋聯配體或電子轉移活化能很低的配合物,它們的機理以外層機理為主。對配體交換反應活性的配合物主要發生橋式機理,橋式機理所需克服的能壘較外層反應機理低很多,因為橋聯配體傳遞電子降低了電子穿透配體外層和水化層的能量。
氧化還原反應中還有兩類反應較特殊:
1.雙電子轉移反應:反應中氧化態的改變為±2,機理為橋式機理的可能性較大。
2.非互補反應:氧化劑與還原劑的價態改變不相等,一般反應機理分幾步進行。
亨利·陶布為配合物的氧化還原反應研究做了很多貢獻,也因此獲得了1983年的諾貝爾化學獎。
套用
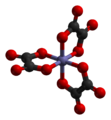
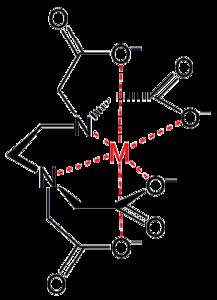 乙二胺四乙酸與金屬離子生成的螯合物。
乙二胺四乙酸與金屬離子生成的螯合物。分析化學中,配合物可用於:
離子的分離:通過生成配合物來改變物質的溶解度,從而與其它離子分離。例如以氨水與AgCl、Hg2Cl2和PbCl2反應來分離第一族陽離子:
AgCl+2NH3→Ag(NH3)2++Cl-
以及利用氨配合物的生成使Zn2+進入溶液:Zn2++4NH3→[Zn(NH3)4]2+。
金屬離子的滴定:例如,定量測定溶液中Fe3+的含量時,指示劑為深紅色的[Fe(phen)3]2+。
掩蔽干擾例子:用生成配合物來消除分析實驗中會對結果造成干擾的因素。比色法測定Co2+時會受到Fe3+的干擾,可加入F-與Fe3+生成無色的穩定配離子[FeF6]3-,以掩蔽Fe3+:
Fe3++6F-→[FeF6]3-
工業生產中:
配位催化:催化反應的機理常會涉及到配位化合物中間體,比如合成氨工業中用醋酸二氨合銅除去一氧化碳,有機金屬催化劑催化烯烴的聚合反應或寡合催化反應,以及不對稱催化於藥物的製備。
制鏡:以銀氨溶液為原料,利用銀鏡反應,在玻璃後面鍍上一層光亮的銀塗層。
提取金屬:例如氰化法提金的步驟中,由於生成了穩定的配離子[Au(CN)2]-,使得不活潑的金進入溶液中:
4Au+8NaCN+2H2O+O2→4Na[Au(CN)2]+4NaOH
也可利用很多羰基配合物的熱分解來提純金屬,例如蒙德法中,鎳的純化利用了四羰基鎳生成與分解的可逆反應:
Ni(CO)4→Ni+4CO。
材料先驅物:氧化鋁微粒及砷化鎵(GaAs)薄膜等的合成。
硬水軟化
生物學中,很多生物分子都是配合物,並且含鐵的血紅蛋白與氧氣和一氧化碳的結合,很多酶及含鎂的葉綠素的正常運作也都離不開配合物機理。常用的癌症治療藥物順鉑,即cis-[PtCl2(NH3)2],可以抑制癌細胞的DNA複製過程,含有平面正方形的配合物構型。乙二胺四乙酸、檸檬酸鈉、2,3-二巰基丁二酸等解毒劑可用於重金屬解毒的機理,常常是它們可與重金屬離子配合,使其轉化為毒性很小的配位化合物,從而達到解毒的目的。
命名法
在命名配位化合物時,一般遵循中文IUPAC命名法,命名規律有:
離子配合物以鹽的形式處理。命名配位單元時,配體在前,不同配體之間以圓點分隔,且最後一個配體與中心原子名稱間要加“合”字。配體的名稱列在右表,其順序主要遵循“先無機後有機”與“先陰離子後中性分子”兩條。配體前要加上配體個數,必要時加圓括弧將配體名稱括起來,以避免歧義。中心原子需在其後標註氧化數,以帶圓括弧的羅馬數字表示。正離子的配合物稱氯化物、硝酸鹽、硫酸鹽等,陰離子的配合物則稱某酸鉀/鈉或某酸。
橋聯配體前要加注μ;ηn則表示配體有n個原子與中心原子鍵結。對於可能產生鍵合異構的配合物,需在配體後註明配位原子。
| 配體 | 作配體時的名稱 | 配合物 | 命名 |
| CO | 羰基 | [NiCl4]2− | 四氯合鎳酸(II)根離子 |
| OH- | 羥(基) | [Cu(NH3)Cl5]3− | 五氯·一氨合銅酸(II)根離子 |
| F- | 氟 | [Cd(en)2(CN)2] | 二氰·二(乙二胺)合鎘(II) |
| PH3 | 膦 | [Co(NH3)5Cl]SO4 | 硫酸一氯·五氨合鈷(III) |
| NO2- | 硝基(-N-) 亞硝酸根(-O-) | Fe2Cl6(氯化鐵二聚體) | 四氯二-μ-氯合二鐵(III) |
| N2 | 雙氮 | (NH4)3[Cr(NCS)6] | 六(硫氰酸根)-N-合鉻酸(III)銨 |
| H2 | 雙氫 | ||
| H- | 氫 |
