
概述
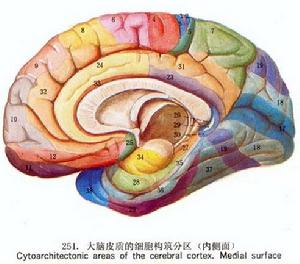 大腦
大腦1960年Levine等首次描述此病,根據遺傳方式、Kell血型,分為常染色體隱性或顯性遺傳的舞蹈病-棘形紅細胞增多症(chorea-acanthocytosisCA)與X-連鎖Mcleod綜合徵兩種類型,其特徵為進行性神經退行性變伴舞蹈樣動作及棘形紅細胞增多。遺傳研究已證實Mcleod綜合徵有X連鎖編碼k蛋白的基因缺失導致定位於紅細胞膜的糖蛋白-KeⅡ抗原表達下降對CA進行基因研究認為其病變位置可能是9q21
以往文獻也曾被稱為伴棘紅細胞增多的家族性肌萎縮性舞蹈病舞蹈病-棘紅細胞增多症家族性神經棘紅細胞增多症等本病以運動障礙(舞蹈症抽動症口下頜運動障礙帕金森綜合徵等)性格改變、進行性智慧型減退、周圍神經病及周圍血棘紅細胞增多為典型的臨床表現
流行病學
尚無權威性的相關的發病率統計學資料NA多見於青春期或成年早期發病年齡8~62歲;病程7~24年存活最長者達33年;男性多於女性男女之比約為1.8∶1
病因
普遍認為神經棘紅細胞增多症(neuroacanthocytosisNA)是一種罕見遺傳病其中以共濟失調為主類型呈常染色體隱性遺傳,以多動為主類型,呈常染色體顯性遺傳,偶有散發病例。也有認為可能是與X染色體基因缺陷相關的性連鎖遺傳病。
發病機制
 神經棘紅細胞增多症
神經棘紅細胞增多症NA的病理改變累及腦(尾狀核嚴重神經元脫失伴膠質細胞增生蒼白球病變較輕)、脊髓(頸髓前角嚴重神經元脫失)、周圍神經(有髓纖維斑片狀脫髓鞘神經元性肌萎縮)等多個部位
屍檢大體標本顯示腦與尾狀核萎縮,側腦室擴大顯微鏡下見紋狀體有小神經元及中等大小神經元缺失廣泛星形細胞反應以尾狀核頭與體萎縮為主神經元數量明顯減少。蒼白球亦有相同改變但程度較輕部分病例丘腦、黑質及脊髓前角有神經元缺失與輕度膠質細胞反應,而腦的其餘部位則相對無改變。個別病例發現腦額葉皮質第3層有不同部位錐體細胞堆積和巨大神經元現象但迄今仍缺乏大樣本病理報告。
臨床表現
 神經棘紅細胞增多症
神經棘紅細胞增多症2.NA最突出的臨床表現是運動障礙,以口面部不自主運動、肢體舞蹈症(酷似HD)最常見。常表現為進食困難,步態不穩,時有自咬唇、舌等。其他運動障礙有肌張力障礙運動不能性肌強直,抽動症,帕金森綜合徵(PDS)等。PDS多見於年輕患者於病程3~7年出現可與上述運動障礙同時出現
3.性格改變和精神症狀亦是其常見症狀;約半數以上患者可有進行性智慧型減退;約1/3患者可出現癲癇發作以強直痙攣性全身發作多見。
4.還可出現周圍神經病EMG顯示失神經支配性肌電圖改變;極少數患者可出現伸跖反射、聽力損害。
5.Haidie等(1991)把NA分為三型:
(1)Bassen-Komzweig綜合徵:又稱無β-脂蛋白血症為常染色體隱性遺傳病臨床表現為棘紅細胞增多、β脂蛋白缺乏、脂肪吸收不良共濟失調視網膜病變可伴肌萎縮、性腺萎縮、弓形足等
(2)Mcleod綜合徵:為X連鎖隱性遺傳病。多於30~40歲發病臨床表現為各種運動障礙常有反射消失、肌病心肌病血清肌酸激酶(CK)活性增高和持續溶血狀態本病的特徵是患者紅細胞表面Kell抗原及xK抗原的抗原性明顯減弱甚或消失。
(3)Levin-Critchley綜合徵:又稱舞蹈病-棘紅細胞增多症。臨床表現與Mcleod綜合徵相似但患者紅細胞表面Kell抗原及xK抗原表達正常血清脂蛋白水平亦在正常範圍。
併發症
約半數以上患者可有進行性智慧型減退,約1/3患者可出現癲癇發作,還可出現周圍神經病極少數患者可出現聽力損害。Mcleod綜合徵可出現肌病、心肌病血清肌酸激酶(CK)活性增高和持續溶血狀態
診斷
 診斷
診斷NA的診斷主要依靠臨床表現及輔助檢查有典型臨床表現周圍血棘紅細胞計數大於3%及血清CK增高者即可診斷
鑑別診斷:
臨床上應注意與慢性進行性舞蹈病(HD)蒼白球黑質紅核色素變性(HSD)及Tourette綜合徵等鑑別。
檢查
 實驗室
實驗室1.普通光鏡檢查可在周圍血中找到棘紅細胞,但只有其計數大於3%才有診斷意義。周圍血中未找到棘紅細胞不能排除NA。
2.紅細胞表面Kell抗原及xK抗原的抗原性減弱或消失是診斷Mcleod綜合徵的重要依據
3.血清β脂蛋白缺如是診斷Bassen-Kormzweig綜合徵的重要依據
4.多數NA患者血清CK活性增高均見於男性患者
其它輔助檢查:1.部分患者EMG檢查表現為失神經支配肌電圖改變。
2.頭顱CT顯示明顯的尾狀核局灶性萎縮Mcleod綜合徵者常有瀰漫性大腦半球萎縮。腦磁共振MRI示雙尾狀核萎縮T1加權像呈低信號,T2加權像與質子密度像顯示尾狀核殼核為略高信號。
3.正電子X線電子計算機斷層掃描(PET)顯示尾狀核殼核大腦皮質的額顳葉,似及丘腦區域腦血流量減少呈低代謝活動
相關檢查:
LDL-膽固醇
甘油三酯
治療
迄今尚無有效治療鎮靜劑如苯巴比妥地西泮、氟哌啶醇對性格、行為障礙,肢體舞蹈症及口面部運動障礙可能有效但易誘發PDS。多巴胺能藥物對PDS可能有所幫助。
預後預防
 藥物
藥物病程7~24年存活最長者達33年;約半數以上患者可有進行性智慧型減退;約1/3患者可出現癲癇發作以強直痙攣性全身發作多見
預防:
由於神經系統遺傳病治療困難,療效不滿意,預防顯得更為重要預防措施包括避免近親結婚,推行遺傳諮詢攜帶者基因檢測及產前診斷和選擇性人工流產等防止患兒出生
相關疾病
 神經
神經>紅斑肢痛症
>維生素E缺乏神經病
>血卟啉病性周圍神經病
>亨廷頓病
>原發性中樞神經系統血管炎
>獲得性免疫缺陷綜合徵的神經系統表現
>神經系統先天性疾病
>顱內肉芽腫性動脈炎
>葉酸缺乏神經病
