
主要病徵
先天性巨指(趾)畸形以手指或足趾軟組織和骨的過度生長為主要特徵,主要包括皮下脂肪、肌腱、神經、脈管、指(趾)骨等全部間葉組織的畸形肥大。但並非所有指(趾)的局限性肥大都稱為先天性巨指(趾)畸形,其他如局部血管瘤、Olllier病、Maffucci綜合徵和Milroy病都可造成指(趾)局部單一組織的增生形成“假性巨指(趾)”。大部分的先天性巨指(趾)畸形為單發症狀,不伴有其他畸形,但在部分罕見的疾病如Proteus綜合徵、Klippel-Trenauany綜合徵、CLOVE綜合徵中,巨指(趾)可作為症狀之一表現出來。
發病機理
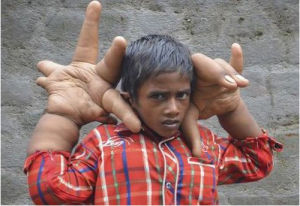
 印度男孩患“巨手症” 手比頭大 每隻重8公斤
印度男孩患“巨手症” 手比頭大 每隻重8公斤 先天性巨指(趾)畸形的發病機理目前仍不明確,該病無明顯遺傳因素,染色體研究未發現異常。Moore於1942年首先提出巨指畸形常伴有周圍神經的病理改變,手術時可見巨指的增生組織內有顯著增長扭曲似曲張靜脈的指神經,因此有假說認為它是一種分布於神經末梢區域的神經誘導的病理改變。Allende認為此病和多發性神經纖維瘤有關,因部分先天性巨指(趾)畸形患者同時伴有皮膚咖啡牛奶斑、脂肪瘤及其他神經纖維瘤病的臨床表現,Gupta亦報導約1/3的先天性巨指畸形患者伴有多發性神經纖維瘤的改變。1987年Keret等曾報導一個同時患先天性巨指和巨趾畸形的病例,其巨指中有神經受累現象,而巨趾中卻沒有發現類似的神經異常,提示巨指和巨趾的發病機理可能並不相同。然而,Syed等認為儘管巨指和巨趾的病理特點可能不同,但其疾病機理一致;局部生長抑制因子缺乏或生長因子過度表達,導致手指或足趾神經損害和脂肪、纖維組織過度增殖。
還有一種觀點認為先天性巨指(趾)畸形是脂肪瘤退變的一種過程,脂肪組織的異常蓄積是本病最明顯的特徵,其脂肪小葉很大,濃密固定且難以去除,肥厚的脂肪還浸潤深層組織。此外,已有巨指(趾)畸形伴小腸脂肪瘤的病例報告。
臨床分型
1967年Barsky在總結相關文獻的基礎上將先天性巨指(趾)畸形分為兩種類型:一種是靜止型,即肥大出生時已伴有,但與其他手指或足趾呈比例關係增長;另一種是進展型,亦為出生伴有,但肥大指(趾)的生長速度遠遠超過正常手指或足趾。部分病人可於出生時並不明顯,而在某一特定時期明顯增大。
顧玉東等提出以巨指體積對先天性巨指畸形進行臨床分型,輕型:為正常指體積的2倍以內;中型:為正常指體積的2-5倍;重型:為正常指體積的5倍以上。
臨床治療
 印度八歲男童長巨手
印度八歲男童長巨手 目前對先天性巨指(趾)畸形尚無統一的治療指南,手術的原則是修復患指(趾)的外形與功能,對於兒童患者應控制其繼續生長。臨床上由於巨指和巨趾的病理學特點不同,且患者對手指和腳趾的外觀和功能要求不同,因此對巨指和巨趾的治療應區別對待。
巨指的治療目的主要是修復畸形手指的外觀,同時強調保留指尖感覺和掌指關節的活動。因此治療必須考慮多方面因素,比如巨指的類型、進展程度和年齡等。輕型者以軟組織(皮膚、脂肪、神經、血管等)切除術為主。中型者以骨組織(骨骺或骨)切除手術為主,重型者以截指為主。而巨趾的治療目的則相對簡單,主要是形成無痛的、美觀的可以舒適穿鞋的腳。巨趾的常用治療方法有:軟組織切除術、骨骺遏止術、截骨術、趾列切除術等。
研究進展
目前關於本病的分子機制研究已有一定進展,2012年Frank等首次發現先天性巨指(趾)畸形的病變組織中多效蛋白(pleiotrophin,PTN)高表達,而PTN作為一種促生長細胞因子能促進局部組織的生長。Rios等採用全基因組測序發現了6名先天性巨指(趾)畸形患者病變組織PIK3CA(R115P)基因突變,而這些突變在患者的血DNA中並未檢出。並據此認為先天性巨指(趾)畸形是由局部組織PI3K/AKT細胞通路過度激活所導致的,且可能與其他過度生長疾病有相同的基因與生化基礎。
病例
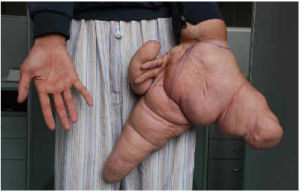 江蘇小伙劉華(化名)在手術前展示巨大的左手和正常的右手
江蘇小伙劉華(化名)在手術前展示巨大的左手和正常的右手 據英國《每日郵報》2014年8月20日報導,印度八歲男孩兒卡里姆(Kaleem)罹患怪病,其雙手比自己的頭還要大,每隻手的重量達到了8公斤,醫生們對此束手無策。
對於這種罕見病,近年來中國媒體也時有報導,最著名的應該是被稱作“世界第一巨手”的江蘇沭陽小伙劉華(化名)。
早在劉華出生後不久,父母就發現他左手的拇指、食指和中指比其他手指明顯粗大。隨著年齡的增長,這3根手指迅速增粗、變長。更讓人意外的是,劉華的整個左手、左臂、左肩和左胸也開始異常增長。截至手術前,他的左手拇指、食指和中指長度分別為26厘米、30厘米和15厘米,整個左臂粗略估計可能重達10公斤。
