
 Rosetta@home
Rosetta@home計算平台
Rosetta@home應用程式和BOINC均支持Microsoft Windows、Linux和蘋果機平台。(BOINC還可在更多平台上運行,如
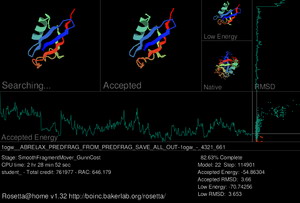 Rosetta@home螢幕保護程式
Rosetta@home螢幕保護程式FreeBSD。)參與Rosetta@home的客戶端需要有一個時鐘速度至少達到500兆赫的中央處理器、400MB空餘硬碟空間、512MB物理記憶體,以及網際網路連線。截至2009年2月9日,Rosetta應用程式的最新版本號是5.98。用戶的BOINC客戶端與位於華盛頓大學的Rosetta@home伺服器端之間使用標準HTTP(80連線埠)進行通信,HTTPS(443連線埠)用於密碼交換。BOINC客戶端使用1043和31416連線埠進行遠程和本地控制,這兩個連線埠可能需要在防火牆中被設定為“解除封禁”才可被使用。包含蛋白質數據的工作單元由伺服器分配給志願者的計算機(客戶端),然後客戶端對所分配得到的任務進行蛋白質預測運算。為了避免重複的預測,每個工作單元會得到一個初始的隨機種子。這使得每個預測具有獨一無二的沿蛋白質能量圖景(energy landscape)的下降軌道。對於給定的蛋白質能量圖景,Rosetta@home的結構預測近似為整體極小值。這個整體極小值代表該蛋白質的能量最佳構造,即它的自然態。
Rosetta@home的圖形用戶界面是一個螢幕保護程式,顯示了當前工作單元進行蛋白質摺疊模擬的情況。螢幕左上方為當前蛋白質鏈正在嘗試的移動(即搜尋的形狀)。緊鄰其右側是最新一個被接受的移動。再往右側分上下兩個小圖,上圖為當前最低能量形狀,下圖為實驗中得到的真實形狀(如果已知)。螢幕中部顯示的是被接受模型的自由能變化曲線。螢幕上方最右側是接受模型的均方根偏差(RMSD)曲線,體現了被接受模型與實驗中真實模型之間的相似度。在自由能變化曲線右側、RMSD曲線下方,使用這兩項結果生成了一個能量/RMSD圖,伴隨著模型的不斷精確。
與所有其他BOINC項目類似,Rosetta@home利用客戶端空閒資源,在後台執行。執行過程可能發生在用戶登錄作業系統之前或之後。當其他應用程式需要時,Rosetta@home會釋放資源,因此不會影響用戶對計算機的正常使用。為了使計算機的功耗或者放熱降到最低,用戶可以自行指定Rosetta@home使用CPU資源的最大比例。此外,Rosetta@home每天最多運行次數,以及更多選項也都可以通過用戶的賬戶選項來進行設定。
Rosetta@home網路所使用的Rosetta軟體最早用Fortran編寫,後改用C++重新編寫,以利於進一步的開發。新版本於2008年2月8日發布,實現了面向對象。Rosetta代碼由Rosetta Commons開發。這個軟體對學術使用免費,對製藥公司則收取費用。
項目意義
 Rosetta@home
Rosetta@home通過一系列的基因組測序計畫,科學家能夠判定許多種在細胞內發揮作用的蛋白質的胺基酸序列或者一級結構。為了更好地了解蛋白質的功能,以及向合理化藥物設計提供輔助,科學家們需要知道蛋白質的三級結構。
蛋白質的三維結構目前主要通過X射線晶體學或核磁共振技術來進行實驗確定。這個過程十分耗時,例如可能花費數星期或者幾個月才能首次研究出如何使一種蛋白質結晶;而成本又非常高,每種蛋白質耗費約10萬美元。更重要的是,發現新序列的速度遠超確定結構的速度:美國國家生物技術信息中心非冗餘蛋白質資料庫中存在的超過740萬個蛋白質序列中,僅有不到5.2萬已被確定結構並被存入蛋白質資料庫中。Rosetta@home的一個主要目標是在顯著降低時間和金錢成本的情況下,預測蛋白質結構,並且達到與現有實驗方法同樣的精度。Rosetta@home還開發了確定膜蛋白(如G蛋白偶聯受體)結構和對接的方法。膜蛋白是現代藥物設計的主要目標,但通過X射線晶體學、核磁共振等傳統技術卻極難獲得其結構。
蛋白質結構預測的進展通過兩年一屆的蛋白質結構預測技術的關鍵測試(CASP)實驗來進行評估。在這項實驗中,來自全球各地的研究人員嘗試從胺基酸序列中得到蛋白質結構。這項實驗有時競爭十分激烈,得分高的小組被認為是最高端蛋白質結構預測研究的事實上的標準制定者。Rosetta@home所基於的Rosetta程式,自1998年的CASP3實驗上開始被使用。在2004年的CASP6上,Rosetta創造了歷史,在它為CASP目標蛋白質T0281提供的模型中,首次生成了接近原子級精度的ab initio蛋白質結構預測。Ab initio不使用結構同源的信息,而必須依賴於序列同源信息以及蛋白質內的模擬物理互動,因此被認為是一類特別難以預測的蛋白質結構。Rosetta@home自2006年CASP7上開始被使用。
在CASP7上,它在每個類別的預測中都成為最好的預測器之一。而高質量的預測需要來自Rosetta@home眾多志願者提供計算資源。不斷增多的計算資源使Rosetta@home能夠對構象空間(一個蛋白質可以被假設具有的可能的形狀)更多的區域進行取樣,根據“黎文索爾佯謬”(Levinthal paradox),採樣數量會隨著蛋白質長度的增長而呈指數增長。
Rosetta@home也被用於蛋白質-蛋白質對接預測。這項預測確定蛋白質複合體結構或者四級結構。這一類型的蛋白質互動作用影響到許多細胞功能,包括抗原-抗體、酶-抑制劑捆綁等。確定這些互動作用在藥物設計中十分關鍵。Rosetta被用於互動作用預測的關鍵測試(CAPRI)實驗。這項實驗評估當前最前沿的蛋白質對接技術,評估模式與CASP類似。Rosetta在這項實驗中得到的結果屬最精確、最完整之一,而志願者提供的計算資源被認為是Rosetta獲得成功的主要因素之一。
2008年初,Rosetta被用來推算設計一種具有在自然界中從未被觀察到功能的蛋白質。這個靈感源自2004年一篇引起關注的、被撤稿的論文,那篇論文中描述了一種蛋白質的推算設計,這種蛋白質與天然的蛋白質相比提高了酶活力。2008年,大衛·貝克的研究組發表了論文,描述了這種蛋白質的製造過程。論文指出Rosetta@home為其提供計算資源,作為這種蛋白質設計方法的一項重要的概念論證。這一類型的蛋白質設計將來可能在藥物設計、綠色化學、生物修復等領域得到套用。
疾病相關研究
除了蛋白質結構預測、對接、設計等基礎研究,Rosetta@home也被用在疾病相關研究。大衛·貝克的Rosetta@home日誌上描述了許多附屬的研究項目。
阿茲海默病
Rosetta套裝軟體當中的一個組件RosettaDesign,被用來精確預測澱粉樣蛋白(amyloidogenic protein)的哪個區域最可能形成澱粉樣纖維。通過獲取蛋白質中的六肽(6個胺基酸長度的片斷),選擇與一個已知能夠形成纖維的六肽相匹配的最低能量結構,RosettaDesign能夠識別出形成纖維可能性為隨機蛋白質兩倍的肽段。在此類研究中,Rosetta@home被用於預測β澱粉樣蛋白的結構。β澱粉樣蛋白是一種能夠形成纖維的蛋白質,被認為會引起阿茲海默病。RosettaDesign一項尚未發表的初步結果設計出了也許可以預防纖維形成的蛋白質,但它能否預防這種疾病尚不得而知。
炭疽病
Rosetta的另一個組件RosettaDock與實驗方法相結合,被用於構造致死因子(lethal factor,LF)、水腫因子(edema factor,EF)和保護性抗原(protective antigen,PA)等三種蛋白質的互動作用模型。這三種蛋白質構成了炭疽病毒素。這個模型精確預測LF與PA之間的對接,幫助確定兩種蛋白質分別有哪些結構域參與到LF-PA複合體的構造中。這項成果最終被套用到改良的炭疽病疫苗的研製。
單純皰疹病毒1型
RosettaDock還被用來構造一種抗體(免疫球蛋白G)與能夠使抗病毒抗體退化的單純皰疹病毒1型(HSV-1)表面蛋白之間的對接模型。RosettaDock預測的蛋白質複合體與極難得到的實驗模型近乎一致。研究人員總結說,這種對接方法有望解決X射線結晶學方法構造蛋白質-蛋白質界面模型所遇到的一些問題。
HIV
作為一項獲得比爾與美琳達·蓋茨基金會1940萬美元資助的研究項目的一部分,Rosetta@home被用於設計人類免疫缺陷病毒(HIV)疫苗。
瘧疾
在與“全球重大衛生挑戰計畫”相關的研究中,Rosetta還被用於運算設計新型的歸巢核酸內切酶蛋白質。這種蛋白質能夠根除甘比亞瘧蚊或者使這種瘧蚊無法傳播瘧疾。由於能夠構建及改變蛋白質-DNA互動作用模型,特別是歸巢核酸內切酶蛋白質等,像Rosetta這一類運算蛋白質設計方法成為基因治療中的一個重要角色。
發展歷史與分支
Rosetta最初是作為ab initio蛋白質結構預測方法,在1998年由貝克實驗室開發;而到目前,這個項目已經形成多個分支,有各自不同的發展和服務方向。Rosetta平台得名於羅塞塔石碑(被用於破譯古埃及文本),因為該平台試圖破譯蛋白質的胺基酸序列的結構“含義”。Rosetta出現7年之後,Rosetta@home項目於2005年10月6日發布。許多參與到Rosetta起步工作的研究生和研究人員已經搬遷到各個不同的大學和研究機構。這使Rosetta項目的不同分支也得到加強。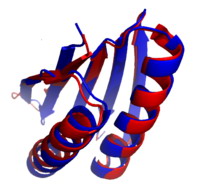 Rosetta設計的TOP7模型
Rosetta設計的TOP7模型RosettaDesign是基於Rosetta的一個蛋白質設計工具。它始於2000年對蛋白質G的摺疊路徑的一項研究。2002年,RosettaDesign被用來設計TOP7。TOP7是一種93個胺基酸長度的α/β型蛋白質,具有在自然界中尚未被發現的整體摺疊結構。這一全新的結構由Rosetta預測,與X射線晶體學確定的結構的之間的RMSD在1.2埃範圍內,是一個精度極高的結構預測。Rosetta和RosettaDesign最早設計和精確預測出這一長度的新型蛋白質,受到廣泛的認可。他們2003年在《科學》雜誌上發表的相關論文已被270多篇論文引用。這項研究的顯著成果TOP7被選為蛋白質資料庫2005年10月的“月度分子”(Molecule of the Month)。這項預測與其X射線晶體學結構的疊合被包含在Rosetta@home的logo設計中。曾在貝克實驗室當博士後、現任北卡羅來納大學教堂山分校助理教授的布賴恩·庫爾曼提供了RosettaDesign的線上服務。
RosettaDock
RosettaDock在2002年CAPRI實驗時作為貝克實驗室的蛋白質-蛋白質對接預測算法被添加到Rosetta軟體套裝中。在那次實驗中,RosettaDock對化膿性鏈球菌毒素A和T細胞受體β鏈的對接做出了高精度預測,對一種豬的α澱粉酶與相應駱駝抗體的複合體做出了中等精度預測。儘管RosettaDock方法只在七種可能中給出兩種可接受精度的預測,這已經足以讓它在那屆CAPRI實驗中名列19個預測方法中的第7位。
RosettaDock的基礎工作主要是由傑弗里·格雷在華盛頓大學期間完成的。後來他搬到約翰·霍普金斯大學接受另一個工作職位。因此,RosettaDock在這之後的開發出現了兩個分支。這兩個分支在側鏈建模、誘捕選擇等方面存在細微差異。儘管存在這些差異,貝克和格雷的方法均在第二次CAPRI實驗中表現出色,在30個小組中分列第5位和第7位。
2006年10月,RosettaDock被集成到Rosetta@home中。這一方法首先僅用蛋白質骨架進行快速、粗略的對接建模,然後進行緩慢的全原子最佳化。在後一階段中,兩個互相作用的蛋白質之間的相對位置以及蛋白質-蛋白質界面的側鏈互動作用同時被最佳化,從而得到最低能量構造。Rosetta@home網路提供的巨大的運算能力,以及骨架靈活性和連線環建模經修訂的摺疊樹表述,使RosettaDock在第三次CAPRI實驗中名列63個預測組中的第6位。
Robetta
Robetta是一個自動化的蛋白質結構預測服務,由貝克實驗室提供,用於非商業性質的ab initio和比較建模。它自2002年起參加CASP實驗,在當屆CASP5的自動化伺服器預測類別中名列前茅。此後,Robetta又參加了CASP6和CASP7,成績比自動化伺服器和人工預測組的平均水平都高。
到CASP6時為止,Robetta構建蛋白質結構模型時採用的方法是,首先用BLAST、PSI-BLAST和3D-Jury搜尋結構同源體,然後通過序列與Pfam資料庫中的結構族的匹配,將目標序列解析為單獨的結構域或者獨立的摺疊單元。下一步,具有結構同源體的結構域則要遵循一個基於模板的模型(即同源建模)協定。在此處,貝克實驗室內部的一個程式K*sync會生成一組序列同源體,其中的每一項由Rosetta的de novo方法建模,產生誘捕(可能的結構)。然後,由低解析度Rosetta能量函式確定的最低能量模型被選為最終的結構預測方案。對於未檢測到結構同源體的結構域,將根據de novo協定,選定生成的誘捕中具有最低能量的模型作為最終的結構預測方案。這些結構域預測方案將被連線在一起,用來研究蛋白質內跨結構域、三級結構級別的互動作用。最後,根據一個蒙特·卡羅構造搜尋協定來構建側鏈貢獻。
在CASP8中,由於Rosetta高解析度全原子最佳化方法的引入,Robetta的性能得到提高。而缺少這一方法被認為是Robetta在CASP7中精度低於Rosetta@home的主要原因。
foldit
2008年5月9日,貝克實驗室接受Rosetta@home用戶關於互動式版本的建議,發布了Foldit。這是一個基於Rosetta平台的線上蛋白質結構預測遊戲。截至2009年1月9日,Foldit的註冊用戶已經接近7.9萬名。這個遊戲賦予用戶一系列的控制功能(如“搖動”、“擺動”、“重建”等),來操縱目標蛋白質的骨架和胺基酸側鏈,以獲得最佳能量構造。用戶能夠以單獨或者集體的形式來進行遊戲,通過改進結構預測方案來獲得積分。用戶還可以通過“決鬥”功能來與其他用戶進行競賽,在20個動作內得到最低能量結構的用戶獲勝。
與類似的分散式計算項目的比較
目前有多個分散式計算項目與Rosetta@home具有類似的研究方向,但研究方法上存在差異。
Folding@home
史丹福大學開發的Folding@home是與蛋白質研究相關的主要分散式計算項目中唯一不使用BOINC平台的Rosetta@home與Folding@home都研究蛋白質錯誤摺疊疾病(如阿茲海默病),但Rosetta@home還進行其他研究,而Folding@home則主要集中於這類研究。Folding@home並不採用基於結構或者基於設計的方法來預測澱粉體行為,而是採用分子動力學方法來構建蛋白質摺疊活動(以及可能的錯誤摺疊和聚合)的模型。換言之,Folding@home的優勢在於模擬蛋白質摺疊活動,而Rosetta@home的優勢則在於蛋白質運算設計以及蛋白質結構和對接的預測。這兩個項目在計算資源和主機分布上也存在顯著差異。Rosetta@home的主機群體基於PC,平均執行速度為78萬億FLOPS;而Folding@home的主機群體包括了PlayStation 3和圖形處理器,平均執行速度達4769萬億FLOPS,大約是Rosetta@home的61倍。
世界公共格線
世界公共格線的子項目人類蛋白質組摺疊項目(HPF)1期和2期均使用Rosetta程式來為不同的基因組添加結構和功能註解。人類蛋白質組摺疊項目的首席科學家裡夏爾·博諾在華盛頓大學貝克實驗室攻讀博士學位期間積極參與了Rosetta的早期開發,但他現在主要使用Rosetta來為生物學家創建資料庫。他的個人網站上設定了關於HPF1、HPF2的信息布告板。
Predictor@home
與Rosetta@home相似,蛋白質結構預測也是Predictor@home的研究重點。Predictor@home還計畫在其分散式計算平台上開發蛋白質設計與對接的新研究領域(採用分子動力學的CHARMM軟體包)。這將使它與Rosetta@home更加相似。進行結構預測時,Rosetta@home使用的是Rosetta程式,而Predictor@home則使用dTASSER方法。
其他
BOINC平台上其他的蛋白質相關分散式計算項目包括QMC@Home、Docking@home、POEM@home、SIMAP和TANPAKU。RALPH@home是Rosetta@home的alpha版本,用來進行新應用程式、工作單元以及更新被添加到Rosetta@home之前的測試工作。RALPH@home也在BOINC平台上運行。
志願者的貢獻
Rosetta@home的研究依賴於大量志願者提供的計算資源。截至2009年1月9日,Rosetta@home的活躍用戶超過4.5萬
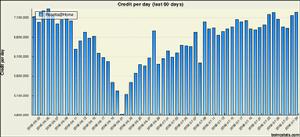 Rosetta@home在CASP8實驗期間的用戶貢獻柱狀統計圖
Rosetta@home在CASP8實驗期間的用戶貢獻柱狀統計圖人,分布在168個國家,總共提供了8.2萬台計算機的空閒資源,使Rosetta@home的平均執行速度達到73萬億FLOPS。
用戶的貢獻通過BOINC積分來度量。一個用戶從某一工作單元得到的積分是他在這個工作單元上產生的“誘餌”數量與所有用戶在該工作單元獲得的積分均值的乘積。Rosetta@home對CPU每秒工作所給的積分低於絕大多數其他BOINC項目。儘管如此,Rosetta@home仍在所有BOINC項目中名列總積分值第5位。
預測的蛋白質結構被提交到CASP實驗的用戶,也在相關的學術出版物中被致謝。而預測出指定工作單元最低能量結構的用戶及其所在隊伍則會在Rosetta@home的主頁上被列為“當日預測者”(Predictor of the Day)。每天還有一名隨機選出的用戶會被列在主頁上,成為“當日用戶”(User of the Day)。
