病理概述
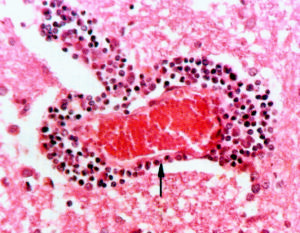 希爾德病
希爾德病病因機制
 希爾德病
希爾德病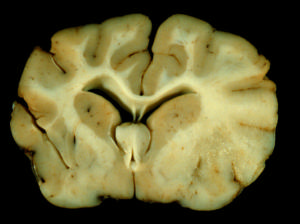 希爾德病
希爾德病臨床表現:
 希爾德病
希爾德病鑑別診斷:
患者臨床表現與急性脫髓鞘性腦病及病毒性腦炎相似病毒性腦炎中以單純皰疹性腦炎最常見,雖然單純皰疹性腦炎呈急性發病精神症狀常見,與本病有相似之處但體溫較高,各種類型的癲癇發作多見;腦脊液多有細胞、蛋白增高而且常見紅細胞CT或MRI可見顳葉和(或)額葉病灶,並且灰白質均可有改變。本病臨床上還易與腎上腺腦白質營養不良(ALD)混淆,ALD為性連鎖遺傳僅累及男性,腎上腺萎縮伴周圍神經受累及NCV異常,極長鏈脂肪酸(VLCFA)含量增高。廣義的彌散性硬化分類中還包括Krabbe球形細胞腦白質營養不良和Greenfield異染性腦白質營養不良等腦白質營養不良性疾病。這組疾病可影響周圍神經也可累及CNS組織腦白質營養不良的臨床特點為進行性視力衰退智慧型減退和痙攣性癱瘓。病理為大片的或多或少兩側對稱性的大腦半球白質破壞每種類型的腦白質營養不良有特異的遺傳性髓鞘脂蛋白代謝的生理缺陷。 希爾德病
希爾德病本病應與以下疾病鑑別:(1)急性播散性腦脊髓炎。為感染和預防接種導致的自身免疫反應性疾病,多於接種或感染後4~14 d起病,可有發熱,突發頭痛、嘔吐等顱高壓征,同時出現腦和脊髓受累症狀,CT或MRI改變似多發硬化,病變位於皮質下白質。(2)多發性硬化:女性多見,40歲左右多發,具有緩解和復發特點的病程。症狀多樣性,病灶好發於腦室周圍、視神經、脊髓、腦幹等。CI1和MRI檢查示病灶的橢圓長徑垂直於側腦室的邊緣,多個並可融合成較大病灶。MRI示病灶呈長Tl長12信號。(3)亞急性多灶性白質腦病。係為乳頭多瘤空泡病毒感染所致進行性脫髓鞘病,多為免疫功能低下患者易患,男性多見。CT早期可無改變,MRI示長Tl、長12信號,少強化,無占位效應,病灶為雙側對稱性的大腦半球皮質下白質局灶或融合成片狀灶,以頂枕葉及額葉多見。(4)腎上腺腦白質營養不良。兒童多見,伴性隱性遺傳,cT或MRI特點為伴有腦萎縮的病灶位於雙側枕葉、常跨越胼胝體底部雙側相連,可向前後發展,且有病變沿傳導束擴張和分布的特點。
病理檢查
 希爾德病
希爾德病主要依據:(1)兒童或青步年發病,亞急性發病逐漸進展病程。(2)智慧型減退,視力下降甚至皮層盲、瘴痛以及精神症狀等表現。(3)影像學示雙側半卵圓中心,以枕葉為主的白質脫鞘改變。(4)長鏈脂肪酸含量正常。(5)排除其它疾病。Cr表現為雙側半球白質廣泛低密度改變=MRI示雙側半球白質長長改變,病灶邊緣界線較清楚,自室營膜下至皮質下,以枕葉為主,雙側可不對稱。率組病例影像學檢查均符合此病的改變,均支持瀰漫性硬化改。
